Mapping Sensitivity in Jaw-Shape Evolutionary Rates with rateMap, Part 1
Source:vignettes/rate-map-jaw-shape-vignette.Rmd
rate-map-jaw-shape-vignette.RmdOverview
The jaw-shape vignette fits one
conservative bifrost search to Paleozoic fish lower-jaw
shape data from Troyer et
al. (2025). It then colors the selected SIMMAP-style tree by fitted
regime-specific rates.
Here we use rateMap() in two steps. First, compute a
reusable "rateMap" object from a list of completed
bifrost searches. Then, call plot() on that
object to render a first-pass equal-weight map, inspect the fitted-rate
range, and redraw the map with explicit near-zero metadata. Because
ordinary bifrost shifts occur at nodes, the default
computed summary is one fitted log-rate per branch.
The same rate-diagnostic controls can be used with a single fitted search; here we introduce them after the first-pass sensitivity map, where the broad fitted-rate range makes the display choice visible.
Methodologically, rateMap() follows the
phytools SIMMAP plotting ecosystem: it is a rate-valued
analogue of densityMap(), and the plot method renders
through plotSimmap() and plotTree(). Details
and acknowledgements are collected near the end of the vignette and in
?plot.rateMap.
This vignette is self-contained for the rateMap()
workflow, while the biological setup comes from the main jaw-shape
analysis. Long-running search chunks are shown for interactive use; run
them once, save the fitted objects, and then use the lighter
rateMap() chunks for summaries and
plot(x, ...) chunks for display. Part 2 continues with IC
weighting, uncertainty views, and same-topology tree samples.

Figure 1. Single-run branch-rate visualization from the
jaw-shape example. This tree shows the fitted branch-rate
categories for one selected bifrost search on the jaw-shape
data; branch colors encode the regime-specific fitted evolutionary rates
from that single model. The multi-run rateMap() workflow
below asks which parts of this single-run pattern persist when the
search threshold changes; subsequent maps use log fitted rates by
default.
Why a Threshold Sweep?
Simulation-based assessment is the primary way to evaluate method-level performance because the generating model, true shift locations, and true rates are known. Simulations ask whether a threshold recovers real shifts, avoids false positives, and behaves well across tree sizes, trait dimensions, and effect sizes. The GIC threshold used in the main jaw-shape analysis follows that logic: values near 20 are conservative, but neighboring thresholds still deserve a dataset-specific sensitivity check.
The sweep here asks a narrower empirical question: does the jaw-shape
rate pattern persist as the shift acceptance threshold becomes more or
less conservative? rateMap() turns repeated
bifrost searches into branchwise summaries that help
distinguish high-rate regions that persist across threshold settings
from features that appear only under permissive settings. This
complements simulation-based model-performance assessment: simulations
benchmark the method, while threshold sweeps diagnose robustness in this
dataset.
We keep min_descendant_tips fixed throughout the sweep.
That argument defines which internal nodes may receive candidate shifts;
changing it would change the candidate-shift universe and the evidence
threshold at the same time. Holding it constant isolates the acceptance
threshold. For the jaw-shape data, min_descendant_tips = 10
also avoids fitting separate high-dimensional covariance structure to
very small clades. That value is a worked-example choice, not a general
default: real analyses may need smaller or larger values depending on
tree size, trait dimensionality, and the smallest clades that should be
allowed to carry their own fitted regime.
Setup
Load the packages used in the jaw-shape workflow.
geomorph::two.d.array() converts the landmark array into a
two-dimensional species-by-trait matrix, matching the setup in the main
jaw-shape vignette.
# Load the search, phylogenetic, and landmark-shape dependencies.
library(bifrost)
library(ape)
library(phytools)
library(geomorph)Load and align the same data used in the jaw-shape vignette.
On first use, the resolver downloads approximately 3 KB for the tree and 245 KB for the landmarks from the maintained GitHub copy of the cited Dryad dataset. Later calls reuse the verified cache. This explicit command checks for an update:
# Refresh the jaw tree only when an updated copy is wanted.
bifrost_example_file("jaw-tree", refresh = TRUE)
# Resolve and read the jaw-shape tree and landmark coordinates.
tree_path <- bifrost_example_file("jaw-tree")
landmark_path <- bifrost_example_file("jaw-landmarks")
fish.tree <- readRDS(tree_path)
landmarks <- readRDS(landmark_path)
# Standardize the tree and align the trait rows to its tip order.
fish.tree <- ladderize(untangle(as.phylo(fish.tree)))
fish.data <- two.d.array(landmarks)
fish.data <- fish.data[match(fish.tree$tip.label, rownames(fish.data)), ]
stopifnot(identical(rownames(fish.data), fish.tree$tip.label))Run the Sensitivity Sweep
The main jaw-shape vignette uses
shift_acceptance_threshold = 20. We include that value and
several neighboring choices. Lower thresholds accept smaller IC
improvements; higher thresholds usually accept fewer shifts.
# Define the acceptance thresholds and minimum candidate-clade size.
threshold_grid <- c(10, 15, 20, 25, 30, 40)
min_tips <- 10Use a helper function so every run differs only in the IC threshold.
Per-shift support weights from searchOptimalConfiguration()
are optional here: set uncertaintyweights_par = TRUE only
if you need those diagnostics for each individual search. They differ
from the fit-level weights used by rateMap(weights = "ic"),
which come from each retained run’s optimal_ic and
IC_used.
Think of rateMap() as the post-processing compute step:
it extracts fitted branch-rate trees and regime-rate parameters, matches
branches across retained runs, and returns a reusable
"rateMap" object. The common compute controls in this
vignette are weights, uncertainty,
summary, and log. Once the object exists, use
plot(x, ...) for display. Most users can stop there. Use
rateMapView(x, ...) only when you want to freeze a display
mapping before printing category tables or reusing the exact same
categories in multiple figures. More specialized controls can be passed
as control = list(...) or built with
rateMapControl(); these are mainly for posterior-tree
samples, interval maps, custom extractors, or large sensitivity
sets.
The sweep is the slowest part of the workflow. The examples below
save the fitted searches to a local cache and reload them for the
lighter rateMap() summaries. Run the sweep chunk once, then
reuse the saved results while exploring display, weighting, and
uncertainty options.
# Wrap the search so threshold is the only setting that changes across runs.
run_jaw_threshold <- function(threshold) {
searchOptimalConfiguration(
baseline_tree = fish.tree,
trait_data = fish.data,
formula = "trait_data ~ 1",
min_descendant_tips = min_tips,
num_cores = 4,
shift_acceptance_threshold = threshold,
uncertaintyweights_par = FALSE,
IC = "GIC",
method = "H&L",
error = TRUE,
plot = FALSE,
store_model_fit_history = FALSE,
verbose = TRUE
)
}
# Run the reproducible threshold sweep and label each retained fit.
set.seed(1)
jaw_threshold_runs <- lapply(threshold_grid, run_jaw_threshold)
names(jaw_threshold_runs) <- paste0("GIC_threshold_", threshold_grid)
# Cache the expensive searches for the lighter mapping examples that follow.
cache_path <- rate_map_cache_path()
dir.create(dirname(cache_path), recursive = TRUE, showWarnings = FALSE)
saveRDS(jaw_threshold_runs, cache_path)If the searches have already been run, load the saved results instead.
# Restore the cached searches and recover their numeric threshold values.
cache_path <- rate_map_cache_path()
if (!file.exists(cache_path)) {
stop("No jaw-shape threshold cache found; run the sweep chunk first.")
}
jaw_threshold_runs <- readRDS(cache_path)
threshold_grid <- as.numeric(sub("GIC_threshold_", "", names(jaw_threshold_runs)))Before mapping rates, inspect how the selected model changes across the sweep.
# Compare selected-model complexity and IC improvement across thresholds.
sweep_summary <- data.frame(
threshold = threshold_grid,
n_shifts = vapply(
jaw_threshold_runs,
function(x) length(x$shift_nodes_no_uncertainty),
integer(1)
),
baseline_ic = vapply(jaw_threshold_runs, `[[`, numeric(1), "baseline_ic"),
optimal_ic = vapply(jaw_threshold_runs, `[[`, numeric(1), "optimal_ic"),
IC_used = vapply(jaw_threshold_runs, `[[`, character(1), "IC_used")
)
sweep_summary$delta_ic <- sweep_summary$baseline_ic - sweep_summary$optimal_ic
sweep_summary| threshold | n_shifts | baseline_ic | optimal_ic | IC_used | delta_ic |
|---|---|---|---|---|---|
| 10 | 19 | -335266.3 | -342617.0 | GIC | 7350.7 |
| 15 | 13 | -335266.3 | -342531.2 | GIC | 7264.9 |
| 20 | 17 | -335266.3 | -342739.4 | GIC | 7473.1 |
| 25 | 14 | -335266.3 | -342455.1 | GIC | 7188.8 |
| 30 | 15 | -335266.3 | -342733.4 | GIC | 7467.2 |
| 40 | 12 | -335266.3 | -342409.2 | GIC | 7142.9 |
threshold
is the shift-acceptance threshold; n_shifts is the number
of retained shifts; baseline_ic and optimal_ic
compare the unshifted and selected models; IC_used records
the criterion; and delta_ic is baseline_ic - optimal_ic.
Accepted shifts are not perfectly monotonic because changing the
threshold can alter the greedy search path, not just trim a fixed list
of candidate shifts.
This table gives a first diagnostic. In this run, the main jaw-shape
threshold (20) gives the lowest GIC among the six retained
searches, closely followed by threshold 30. Those two model
outcomes dominate the IC-weighted rate map in Part 2.
Equal-Weight Rate Map
A simple first diagnostic gives every threshold run equal weight. This asks whether the same branch log-rate pattern appears across the threshold settings considered here.
By default, rateMap() follows bifrost’s
branch-level framing: shifts occur at nodes, so each branch receives one
summarized fitted rate. With the default log = TRUE,
across-run means are mean log-rates, equivalent to the log of a weighted
geometric mean on the original rate scale.
plot(x, ...) then colors those branch summaries as
ordered log-rate categories. In multi-run summaries, these categories
are plotting bins for summarized branch values; they should not be read
as newly inferred bifrost regimes. For branch summaries,
rate_categories reports how many branches fall into each
displayed bin and summarizes the plotted branch values assigned to that
bin.
Branch maps also carry rate diagnostics for near-zero or isolated
high-tail rates. These diagnostics add rate_flag metadata
without removing branches or changing fitted values. By default, no
near-zero rule is applied. Supplying zero_floor activates
the manual floor rule; using method = "tail_cluster"
requests the data-driven lower-tail diagnostic.
That conservative default matters because bifrost can
infer very low but positive fitted rates for clades with little observed
trait variation. Leaving those branches unflagged is not wrong; it shows
the fitted branch summaries directly. If you have a biologically or
numerically meaningful threshold in mind, supply zero_floor
to mark rates below that value as effectively zero for display.
rateMap() leaves that choice explicit rather than guessing
a universal cutoff.
Start with the default map and inspect its range before deciding how to display it.
# Compute the unflagged equal-weight map as an initial diagnostic.
jaw_rate_map_equal_first <- rateMap(
jaw_threshold_runs,
weights = "equal",
uncertainty = TRUE,
progress = TRUE
)
# Freeze a common categorical display before inspecting its fitted-rate range.
jaw_rate_map_equal_first <- rateMapView(
jaw_rate_map_equal_first,
palette = "Viridis",
legend_title = "Mean log fitted rate"
)
# Inspect both the map summary and its rate diagnostics.
jaw_rate_map_equal_first
jaw_rate_map_equal_first$rate_diagnostics#> rateMap object
#> - fits: 6
#> - summary: branch
#> - target: first
#> - check: full
#> - weights: equal
#> - uncertainty: TRUE
#> - color mode: category
#> - plotted value: value
#> - value range: -24.2 to -11.41
#> - rate flags: 0 near-zero, 0 high-outlier (method: none)
#> - fold range: 361384 full, 361384 regular
#> - sd range: 0.05957 to 5.979| diagnostic | value |
|---|---|
| method | none |
| near-zero floor | none |
| regular branches | 170 |
| near-zero branches | 0 |
| high-outlier branches | 0 |
| minimum fitted log-rate | -24.2 |
| minimum regular fitted log-rate | -24.2 |
| maximum fitted log-rate | -11.41 |
| full fold range | 361384 |
| regular fold range | 361384 |
The first pass is a useful warning rather than a final display
choice. The branch summaries run from about -24.2 to
-11.4 on the log-rate scale, which corresponds to original
fitted rates of about 3.08e-11 to 1.11e-5. A
361,000-fold range is possible under the fitted model, but
it is biologically hard to interpret as ordinary rate variation. That is
the reason to inspect the lower tail and, when appropriate, display the
effectively zero-rate branches as a separate diagnostic category. The
default plot legend spans the prettier category boundaries, here
-26 to -10, so its lower limit is a bin edge
rather than the observed minimum branch summary.
# Draw the default map before applying a near-zero diagnostic.
plot(
jaw_rate_map_equal_first,
type = "arc",
show_tip_labels = FALSE,
legend_fsize = 0.9,
arc_height = 0.5
)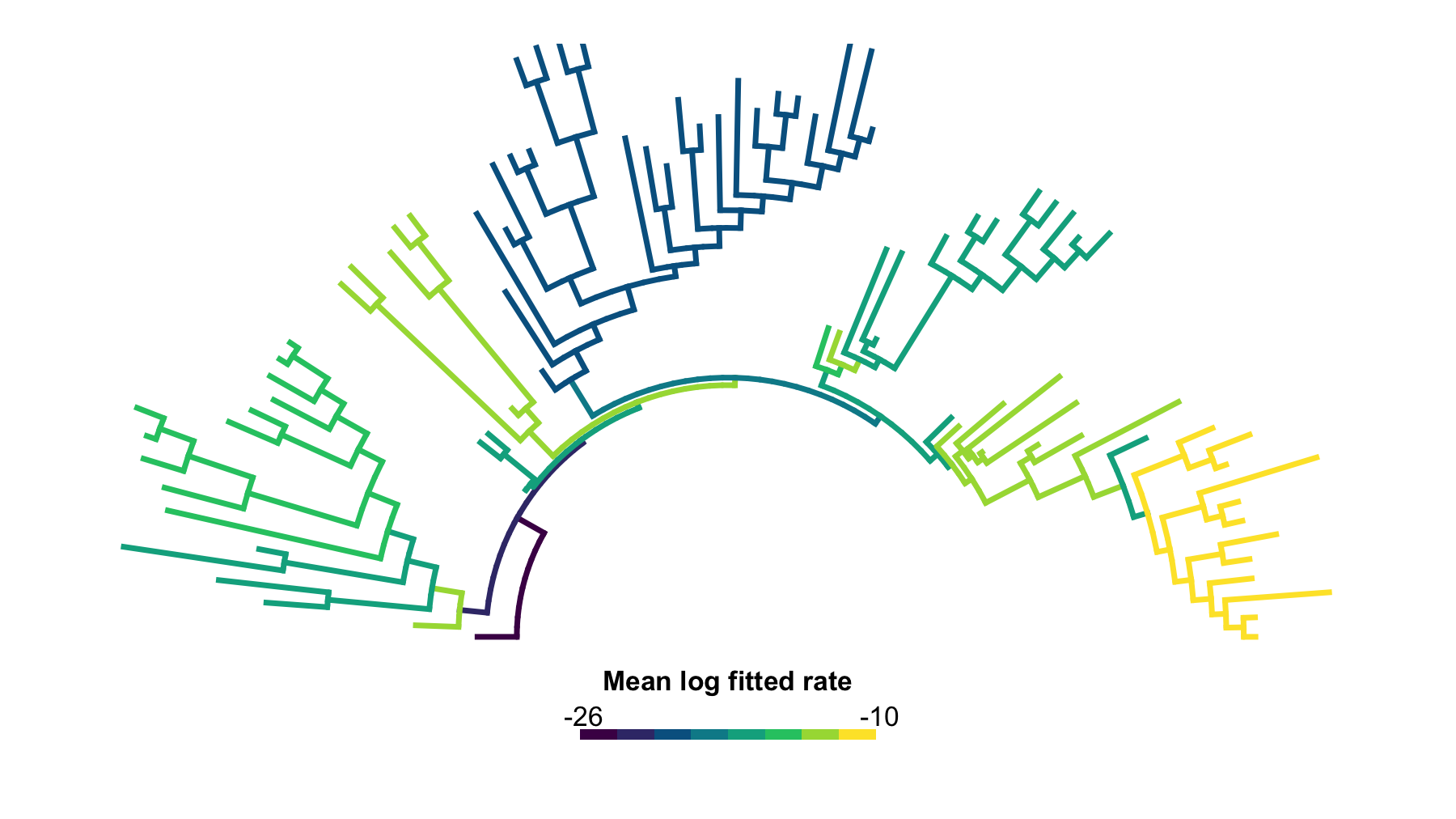
Figure 2. Default equal-weight branch-category rate map. This first-pass arc tree shows the unmodified default display: all branches are binned into the ordinary ordered log-rate categories, with no manual near-zero flagging.
In this jaw-shape example, the lowest-rate branches form a broader lower-tail cluster that deserves an explicit diagnostic display choice.
The second pass keeps the same fitted values but uses the
tail_cluster diagnostic. This Otsu-style rule, adapted from
Otsu’s histogram thresholding criterion (Otsu 1979), works
on log-rates, chooses the guarded two-class split that maximizes
separation between the tail and the remaining branches, and only accepts
it when the lower cluster is separated from the regular branches and
greatly reduces the regular fold range.
# Recompute the equal-weight map with separated lower-tail branches flagged.
jaw_rate_map_equal <- rateMap(
jaw_threshold_runs,
weights = "equal",
uncertainty = TRUE,
progress = TRUE,
control = rateMapControl(
rate_flags = rateMapRateFlags(method = "tail_cluster")
)
)
# Apply a categorical Viridis view to the regular-rate branches.
jaw_rate_map_equal <- rateMapView(
jaw_rate_map_equal,
palette = "Viridis",
legend_title = "Mean log fitted rate"
)
# Inspect the adjusted map and the resulting display categories.
jaw_rate_map_equal
jaw_rate_map_equal$rate_categoriesAfter flagging, the grey near-zero category sits outside the ordered
palette. The Viridis colors are regenerated for the remaining regular
categories, so the unflagged branches use the full low-to-high color
range. The legend still spans the full plotted data range and marks the
cluster cutoff, here about -18.39, between the highest
near-zero branch (-19.79) and the first regular branch
(-16.99). In this example the first occupied regular
automatic bin starts at -17; use explicit
category_breaks if you want the display anchored to a
particular numeric cutoff.
#> rateMap object
#> - fits: 6
#> - summary: branch
#> - target: first
#> - check: full
#> - weights: equal
#> - uncertainty: TRUE
#> - color mode: category
#> - plotted value: value
#> - value range: -24.2 to -11.41
#> - rate flags: 54 near-zero, 0 high-outlier (method: tail_cluster)
#> - fold range: 361384 full, 265 regular
#> - sd range: 0.05957 to 5.979The category table and diagnostic table should be read together: the left table shows how branch summaries are binned for display, and the right table records the near-zero rule that created the grey category.
| rate_category | n_branches | value_mean |
|---|---|---|
| near-zero | 54 | -21.51 |
| -17 to -16 | 40 | -16.38 |
| -16 to -15 | 4 | -15.1 |
| -15 to -14 | 18 | -14.96 |
| -14 to -13 | 16 | -13.4 |
| -13 to -12 | 16 | -12.37 |
| -12 to -11 | 22 | -11.41 |
| diagnostic | value |
|---|---|
| method | tail_cluster |
| tail-cluster log cutoff | -18.39 |
| tail-cluster log gap | 2.803 |
| tail-cluster fold reduction | 1364 |
| tail-cluster branch fraction | 0.3176 |
| regular branches | 116 |
| near-zero branches | 54 |
| high-outlier branches | 0 |
| minimum fitted log-rate | -24.2 |
| minimum regular fitted log-rate | -16.99 |
| maximum fitted log-rate | -11.41 |
| full fold range | 361384 |
| regular fold range | 265 |
The near-zero category is metadata, not a data filter. The full
fitted-rate range is still available in
jaw_rate_map_equal$intervals, but the ordinary rate bins
and the visual color scale now describe the remaining regular branches
rather than stretching across a numerical floor.
# Draw the adjusted map with near-zero branches outside the ordered palette.
plot(
jaw_rate_map_equal,
type = "arc",
show_tip_labels = FALSE,
legend_fsize = 0.9,
arc_height = 0.5
)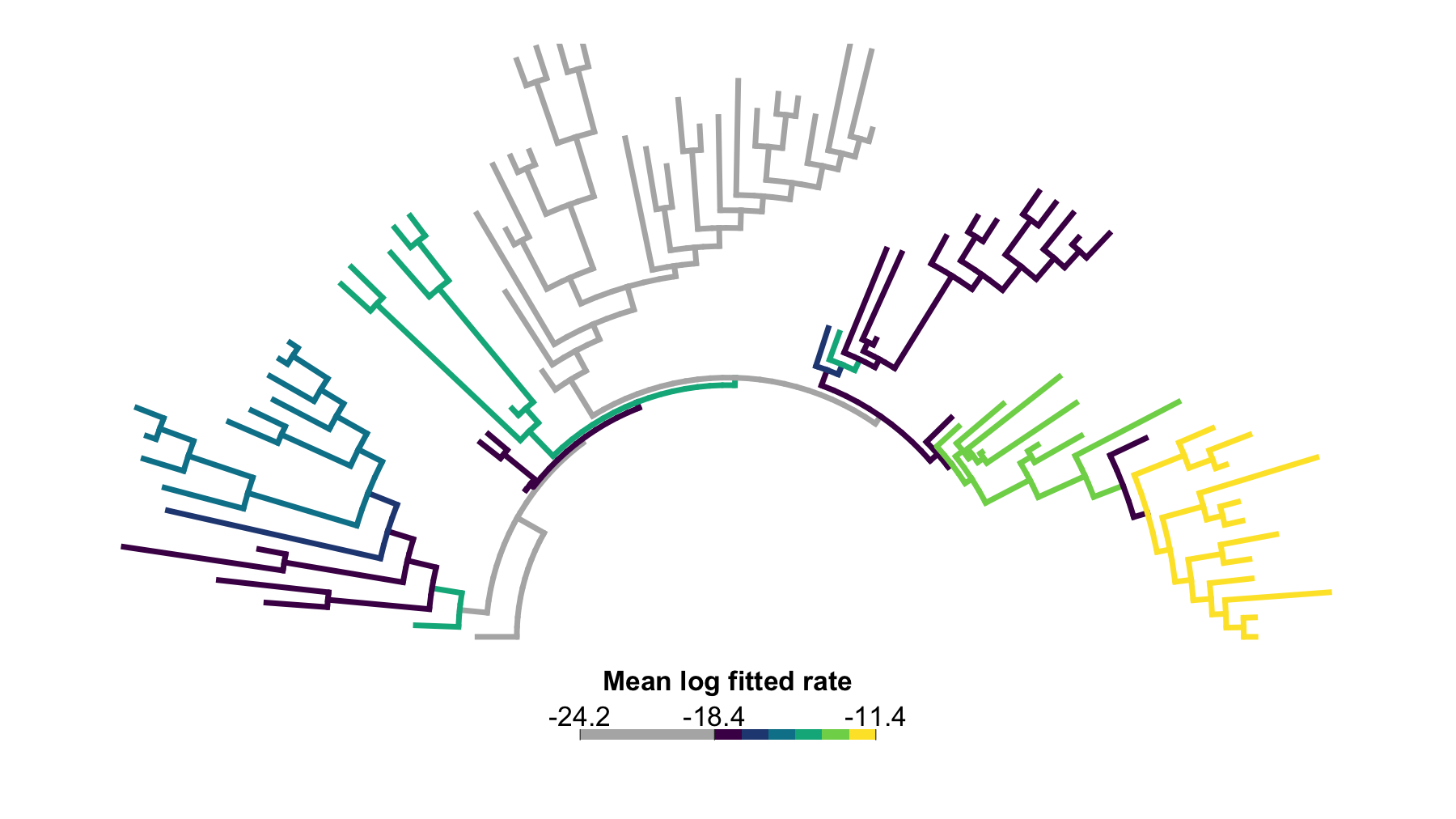
Figure 3. Equal-weight branch-category rate map after near-zero flagging. This arc tree summarizes the six GIC-threshold searches by giving each completed search the same weight. Branches in the separated lower log-rate cluster are shown as a separate grey category; remaining regular branches are colored by ordered categories of their mean fitted branch log-rate.
Each threshold run contributes one sixth of the branch log-rate summary. The highest mean log-rate region occurs in the lungfish-centered part of the tree, including branches descending toward Griphognathus, Holodipterus, Rhynchodipterus, and related taxa.
Interpret this map as a sensitivity average, not as a posterior probability map. Branches with consistently high log-rate values are repeatedly assigned to faster regimes across the threshold sweep; intermediate categories may reflect either genuinely moderate rates or a mixture of low- and high-rate assignments across thresholds.
If you want tighter control over the display categories after this diagnostic step, adjust the plotting bins directly.
# Compare automatic, equal-width, and explicit numeric category controls.
plot(jaw_rate_map_equal, n_categories = 5)
plot(jaw_rate_map_equal, n_categories = 5, category_bin_method = "equal")
plot(jaw_rate_map_equal, category_breaks = c(-17, -15, -13, -11))When manual numeric category_breaks define unequal
intervals, the category legend uses proportional segment widths so the
displayed bin geometry follows the numeric rate scale.
Practical Takeaways
- Holding
min_descendant_tips = 10fixed keeps the candidate-node filter constant whileshift_acceptance_thresholdchanges. - Equal weights summarize robustness across selected threshold settings.
- The first-pass rate diagnostics are part of the workflow: inspect the fitted-rate range before accepting the default color scale.
- The default diagnostic pass applies no near-zero rule; manual floors and broader tail structure should be requested explicitly.
-
method = "tail_cluster"is display metadata, not a data filter. It uses an Otsu-style two-class split to mark a separated lower log-rate cluster while preserving fitted values in the output object. - In
plot(x, ...),n_categoriessets the target number of automatic category bins;category_bin_method = "equal"andcategory_breaksgive more direct bin control; numeric manual breaks also set proportional category legend segment widths;ncolorscontrols continuous ramps.
Next Steps
Part 2 continues from this adjusted equal-weight map and asks how the interpretation changes under IC weighting, across-threshold uncertainty maps, same-topology tree samples, original-scale rates, and branch-level diagnostic tables.
Acknowledgements and Provenance
The core idea is adapted from phytools::densityMap():
summarize mapped histories on a shared tree and display the summary as a
colored phylogeny. rateMap() changes the mapped quantity
from posterior state probability to fitted branch-rate summaries from
completed bifrost searches.
The drawing layer uses phytools::plotSimmap(),
with phytools::plotTree()
for outline and base-tree passes. plot.rateMap() forwards
tree-layout controls such as type, fsize,
ftype, lwd, mar,
direction, offset, xlim,
ylim, underscore, and arc_height;
rate-map controls such as palette, color_mode,
n_categories, category_breaks,
legend_title, and near-zero diagnostics are handled by
rateMap() before the colored tree is drawn.
References
- Clavel, J., Aristide, L., and Morlon, H. (2019). A Penalized Likelihood Framework for High-Dimensional Phylogenetic Comparative Methods and an Application to New-World Monkeys Brain Evolution. https://doi.org/10.1093/sysbio/syy045
- Otsu, N. (1979). A threshold selection method from gray-level histograms. IEEE Transactions on Systems, Man, and Cybernetics, 9(1), 62-66. https://doi.org/10.1109/TSMC.1979.4310076
- Paradis, E., and Schliep, K. (2019). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics, 35, 526-528. https://doi.org/10.1093/bioinformatics/bty633
- Revell, L. J. (2013). Two new graphical methods for mapping trait evolution on phylogenies. Methods in Ecology and Evolution, 4, 754-759.
- Revell, L. J. (2014). Graphical methods for visualizing comparative data on phylogenies. Chapter 4 in Modern Phylogenetic Comparative Methods and Their Application in Evolutionary Biology: Concepts and Practice, 77-103.
- Revell, L. J. (2024). phytools 2.0: an updated R ecosystem for phylogenetic comparative methods (and other things). PeerJ, 12, e16505. https://doi.org/10.7717/peerj.16505
- Troyer, E. M., et al. (2025). Macroevolutionary role reversals in the earliest radiation of bony fishes. https://doi.org/10.1016/j.cub.2025.08.008
Software Used in This Vignette
-
bifrostfor shift searches andrateMap()summaries. -
geomorphfor converting landmark arrays to species-by-trait matrices. -
apeandphytoolsfor phylogenetic data structures, SIMMAP trees, and plotting.