Mapping Sensitivity in Jaw-Shape Evolutionary Rates with rateMap, Part 2
Source:vignettes/rate-map-jaw-shape-part-2-comparisons.Rmd
rate-map-jaw-shape-part-2-comparisons.RmdOverview
Part 1 built the core equal-weight rateMap() workflow:
load a cached threshold sweep, inspect the first-pass fitted-rate range,
and then display effectively zero-rate branches with explicit near-zero
metadata. This second part starts from the same cached jaw-shape sweep
and explores the extensions that are useful after the main map is
stable.
As in Part 1, plotted rate categories are display bins over fitted
branch summaries, not newly inferred bifrost regimes. Rate
flags are diagnostic metadata used for display; they do not remove
branches or change fitted values.
Read Part 1: Workflow and near-zero diagnostics first if you want the setup and main equal-weight map.
Setup
Load the packages needed for the comparison and diagnostic views.
# Load the package and phylogenetic utilities used in the comparisons.
library(bifrost)
library(ape)
library(phytools)Load the reference tree used for plotting and same-topology examples.
This part requests only the approximately 3 KB tree from the maintained GitHub copy of the cited Dryad dataset. The first call downloads and verifies it; later calls reuse the verified cache. This explicit command checks for an update:
# Refresh the jaw tree only when an updated copy is wanted.
bifrost_example_file("jaw-tree", refresh = TRUE)
# Load and standardize the reference phylogeny from Part 1.
tree_path <- bifrost_example_file("jaw-tree")
fish.tree <- readRDS(tree_path)
fish.tree <- ladderize(untangle(as.phylo(fish.tree)))Load the cached threshold sweep created in Part 1.
# Restore the Part 1 threshold sweep and recover its numeric thresholds.
cache_path <- rate_map_cache_path()
if (!file.exists(cache_path)) {
stop("No jaw-shape threshold cache found; run the Part 1 sweep first.")
}
jaw_threshold_runs <- readRDS(cache_path)
threshold_grid <- as.numeric(sub("GIC_threshold_", "", names(jaw_threshold_runs)))Reconstruct the adjusted equal-weight object from Part 1 so uncertainty maps and branch-level diagnostics in this part have the same Otsu-style tail-cluster near-zero metadata, adapted from Otsu’s histogram thresholding criterion (Otsu 1979).
# Rebuild the adjusted equal-weight map used as the uncertainty reference.
jaw_rate_map_equal <- rateMap(
jaw_threshold_runs,
weights = "equal",
uncertainty = TRUE,
progress = TRUE,
control = rateMapControl(
rate_flags = rateMapRateFlags(method = "tail_cluster")
)
)
# Reapply the same categorical display mapping used in Part 1.
jaw_rate_map_equal <- rateMapView(
jaw_rate_map_equal,
palette = "Viridis",
legend_title = "Mean log fitted rate"
)IC-Weighted Rate Map
An IC-weighted map asks a different question: which log-rate pattern is emphasized when lower-GIC threshold outcomes contribute more heavily?
Use this view when the threshold-generated fits form a small set of
comparable model outcomes. All retained runs must use the same IC
family, so do not mix GIC and BIC runs in a single
weights = "ic" map. If one threshold dominates the IC
weights, the result will resemble that single model more than an
across-threshold robustness summary. In a threshold sweep, this can
favor a more permissive fitted configuration, so read the map as a
model-outcome weighting over retained fits rather than as a formal
threshold-selection rule.
# Weight completed threshold searches by their relative GIC support.
jaw_rate_map_ic <- rateMap(
jaw_threshold_runs,
weights = "ic",
uncertainty = TRUE,
progress = TRUE,
control = rateMapControl(
rate_flags = rateMapRateFlags(method = "tail_cluster")
)
)
# Freeze the IC-weighted categorical view for tables and plotting.
jaw_rate_map_ic <- rateMapView(
jaw_rate_map_ic,
palette = "Viridis",
legend_title = "IC-weighted mean log fitted rate"
)
# Inspect the fitted map, fit-level weights, and display categories.
jaw_rate_map_ic
jaw_rate_map_ic$weight_table
jaw_rate_map_ic$rate_categories#> rateMap object
#> - fits: 6
#> - summary: branch
#> - target: first
#> - check: full
#> - weights: ic
#> - uncertainty: TRUE
#> - color mode: category
#> - plotted value: value
#> - value range: -25.79 to -11.36
#> - rate flags: 54 near-zero, 0 high-outlier (method: tail_cluster)
#> - fold range: 1857530 full, 714 regular
#> - sd range: 0.005051 to 0.665| input_index | weight | ic | IC_used |
|---|---|---|---|
| 3 | 0.952 | -342739.4 | GIC |
| 5 | 0.04798 | -342733.4 | GIC |
| 1 | 2.498e-27 | -342617.0 | GIC |
| 2 | 5.760e-46 | -342531.2 | GIC |
| 4 | 1.761e-62 | -342455.1 | GIC |
| 6 | 1.887e-72 | -342409.2 | GIC |
input_index identifies the
retained search in the input list; weight is the normalized
IC weight used by rateMap(); ic is the
selected model’s IC value; and IC_used records the
criterion used to compute comparable weights. In this sweep, inputs 3
and 5 carry nearly all of the IC weight, while the other retained runs
have effectively zero weight.
| rate_category | n_branches | value_mean | value_median | value_sd | total_branch_length |
|---|---|---|---|---|---|
| near-zero | 54 | -21.94 | -21.67 | 1.113 | 388.1 |
| -18 to -17 | 8 | -17.93 | -17.93 | 0 | 107.4 |
| -17 to -16 | 6 | -16.23 | -16.1 | 0.1875 | 33.21 |
| -16 to -15 | 48 | -15.37 | -15.08 | 0.3215 | 312.1 |
| -15 to -14 | 0 | NA | NA | NA | 0 |
| -14 to -13 | 12 | -13.26 | -13.26 | 0 | 110.5 |
| -13 to -12 | 20 | -12.36 | -12.3 | 0.1794 | 132.2 |
| -12 to -11 | 22 | -11.36 | -11.36 | 0 | 126.6 |
rate_category is the displayed interval or diagnostic
class; n_branches counts branches assigned to the bin;
value_mean, value_median, and
value_sd summarize plotted branch log-rates; and
total_branch_length gives the summed branch length in that
displayed bin. The full generated category table remains available as
rate_map_ic_categories.csv.
# Draw the IC-weighted map with near-zero branches shown separately.
plot(
jaw_rate_map_ic,
type = "arc",
show_tip_labels = FALSE,
legend_fsize = 0.9,
arc_height = 0.5
)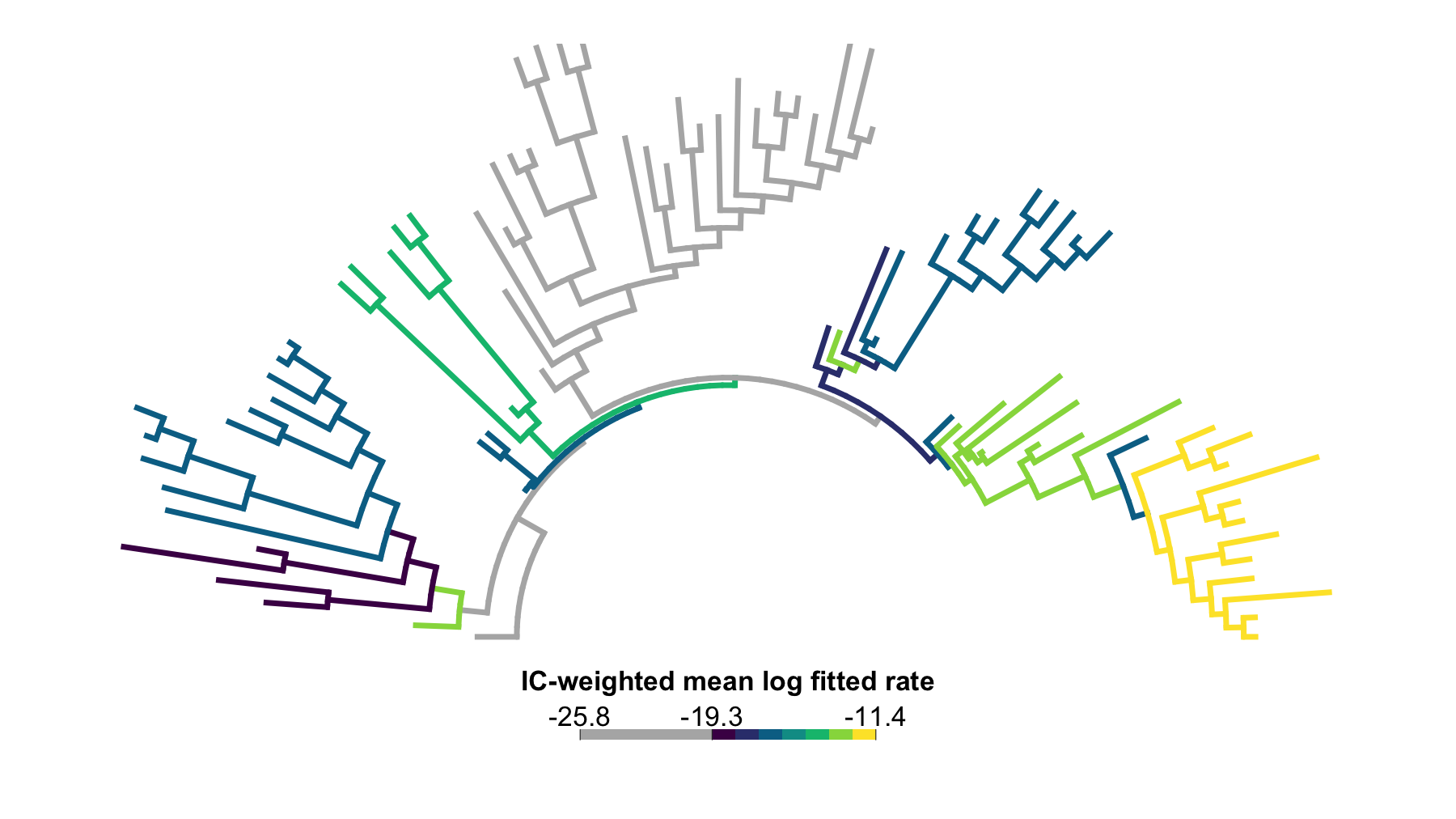
Figure 1. IC-weighted branch-category rate map after near-zero flagging. This arc tree summarizes the same threshold sweep after weighting each completed search by its relative GIC support. Branches in the separated lower log-rate cluster are shown as a separate grey category; remaining regular branches are colored by ordered categories of the IC-weighted mean fitted log-rate.
Here the IC weights concentrate on thresholds 20 and
30, with threshold 20 carrying most of the
weight. The weighting choice does not change the log-rate scale;
weights = "ic" changes only the fit-level weights in the
weighted mean log-rate. The IC-weighted map is visually similar to the
equal-weight map because the dominant lower-GIC searches preserve the
same broad branch-rate structure. The maps are not identical: branch
values are recomputed under IC weights, and some branches shift color
categories, but the main high-rate regions remain stable. Appendix E
shows the largest branch-level differences between weighting
schemes.
Mapping Uncertainty Across Thresholds
With uncertainty = TRUE, rateMap() stores
run-level log-rate values and adds branch-level summaries: weighted
mean, weighted median, standard deviation, quantiles, highest-density
interval bounds, and coefficient of variation. The highest-density
interval contains 95% of the weighted run-level values by default; set
control = list(highest_density_interval_prob = ...) to
change that probability. For equal weights, sd is the
ordinary sample standard deviation across runs; for IC or custom
weights, it is computed from the normalized weighted variance. In this
threshold sweep, these columns summarize variation across fitted
searches. They do not measure landmark error, posterior tree
uncertainty, or uncertainty inside the original fitted models. They also
do not use the per-shift support weights that bifrost can
compute inside an individual search; the weights here are the fit-level
weights supplied to rateMap().
For the equal-weight six-run map shown here, the default 95% highest-density interval is deliberately conservative and effectively spans all retained run-level values for each branch. With denser run stacks, the same column behaves more like the familiar shortest interval containing the requested probability mass; for small sensitivity sweeps, interpret it as a range-like stability diagnostic.
You can recolor the same object by any compatible numeric column. For
uncertainty summaries, continuous color ramps are often easier to read
than discrete categories. The plotted summary table can still be sorted
by sd or highest_density_interval_width for
reporting, but the tree views are the most direct way to see where
threshold sensitivity sits on the phylogeny. For Figures 2 and 3, color
encodes uncertainty rather than rate magnitude; darker branches are more
stable across the threshold sweep, while lighter branches vary more
across runs. As a first example, plot the across-threshold standard
deviation:
# Recolor the equal-weight map by across-threshold standard deviation.
plot(
jaw_rate_map_equal,
value = "sd",
type = "arc",
show_tip_labels = FALSE,
palette = "Inferno",
color_mode = "continuous",
legend_fsize = 0.9,
arc_height = 0.5
)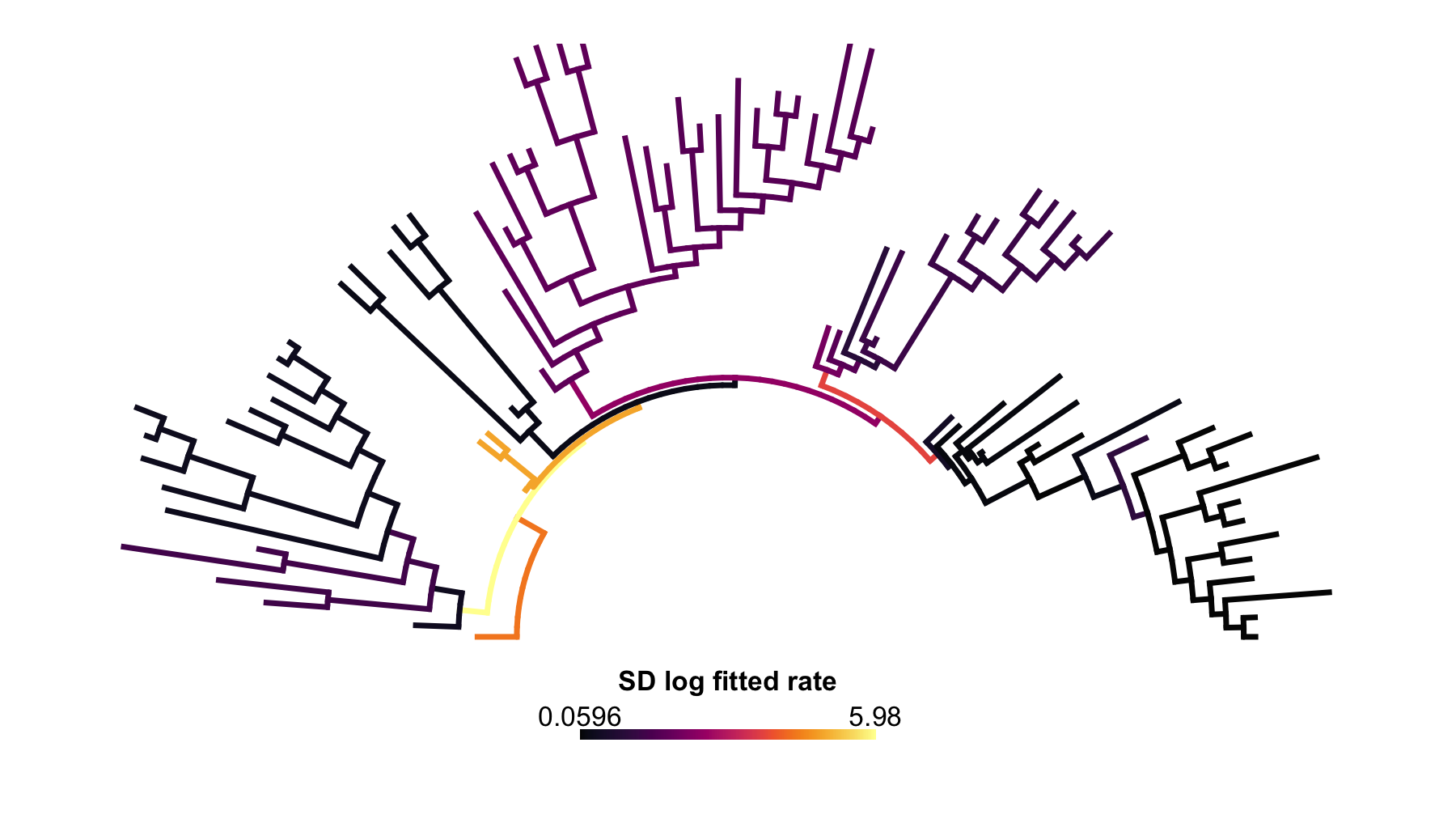
Figure 2. Across-threshold standard deviation. This
uncertainty map recolors the equal-weight rateMap() object
by the standard deviation of fitted branch log-rates across the six
threshold searches. Larger values mark branches whose inferred rates
change more across the threshold sweep, while smaller values mark
branches with more stable rate assignments.
Or plot the highest-density, or shortest weighted, interval width:
# Recolor the map by the 95% shortest-interval width across thresholds.
plot(
jaw_rate_map_equal,
value = "highest_density_interval_width",
type = "arc",
show_tip_labels = FALSE,
palette = "Plasma",
color_mode = "continuous",
legend_fsize = 0.9,
arc_height = 0.5
)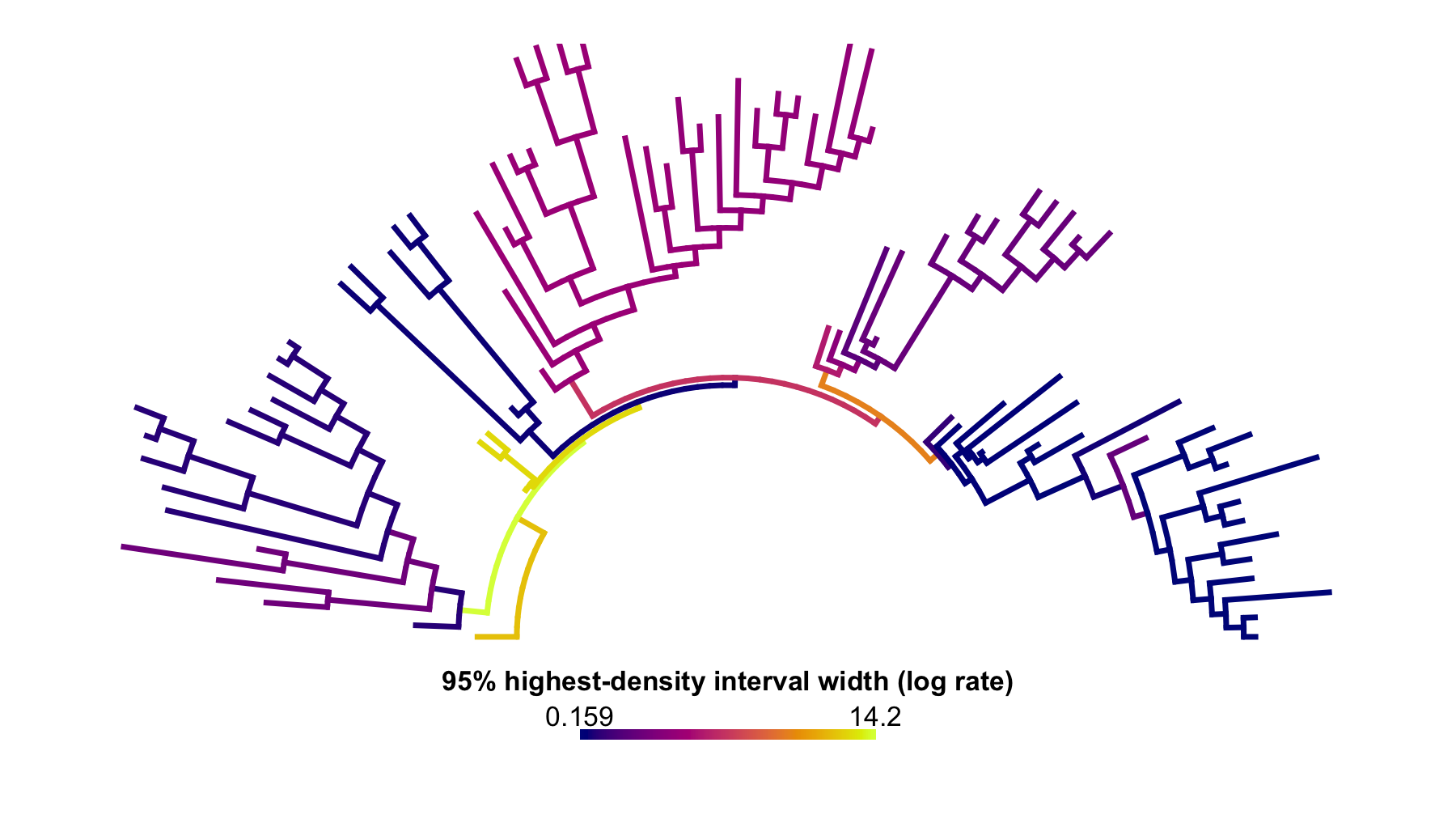
Figure 3. Across-threshold 95% shortest-interval width.
This uncertainty map recolors the equal-weight rateMap()
object by the width of the highest-density, or shortest weighted,
interval containing 95% of the run-level log-rate values for each
branch. Wider intervals indicate less stable fitted rate assignments
across threshold searches; narrower intervals indicate stronger
agreement among runs.
In this example dataset, the strongest uncertainty signal partly
overlaps the near-zero diagnostic. A compact way to check that overlap
is to mark branches in the upper quartile of
highest_density_interval_width, then ask whether near-zero
branches are overrepresented in that high-uncertainty set.
# Flag near-zero branches and branches in the upper uncertainty quartile.
rate_intervals <- jaw_rate_map_equal$intervals
near_zero <- rate_intervals$is_near_zero %in% TRUE
high_interval_width <- rate_intervals$highest_density_interval_width >=
quantile(rate_intervals$highest_density_interval_width, 0.75, na.rm = TRUE)
# Count near-zero branches in the full and high-uncertainty branch sets.
branch_set_n <- c(
nrow(rate_intervals),
sum(high_interval_width)
)
near_zero_n <- c(
sum(near_zero),
sum(near_zero & high_interval_width)
)
# Report the overlap as counts and percentages.
uncertainty_overlap_summary <- data.frame(
branch_set = c(
"all branches",
"high-uncertainty branches"
),
branches = branch_set_n,
near_zero_branches = near_zero_n,
near_zero_percent = sprintf("%.1f%%", 100 * near_zero_n / branch_set_n)
)
uncertainty_overlap_summary| branch_set | branches | near_zero_branches | near_zero_percent |
|---|---|---|---|
| all branches | 170 | 54 | 31.8% |
| high-uncertainty branches | 44 | 34 | 77.3% |
highest_density_interval_width. The near-zero
fraction is higher in that set than across all branches.
In this sweep, near-zero branches are enriched among high-uncertainty
branches: 31.8% of all branches are near-zero, compared
with 77.3% in the upper quartile of shortest-interval
width. This overlap is diagnostic, not redundant: near-zero flags
summarize mean fitted branch rate, while uncertainty columns summarize
across-threshold variation. Regular branches can still be
high-uncertainty when their rates move toward or away from the lower
tail.
High mean log-rate with low across-threshold uncertainty is the strongest visual cue for a robust fast-rate region. High mean log-rate with high uncertainty is more fragile: it may depend on the threshold setting, or it may identify a branch whose regime assignment changes across the sweep.
The plotted summary object still stores one row per branch under the
default summary = "branch"; Appendix D shows how to sort
those rows for reporting.
Same-Topology Tree Samples
The same machinery can summarize fitted rates across a posterior
sample of time-calibrated trees when the topology is fixed but
divergence times and branch lengths vary. For example, a researcher
might fit bifrost to a set of posterior chronograms from a
divergence-time analysis, then ask how the fitted branch-rate pattern
can be summarized on one reference tree.
In that case, provide an explicit target_tree and relax
the tree check from identical branch lengths to matching topology with
control = list(check = "topology"). The target can be an
MCC tree, a posterior mean chronogram, or another biologically preferred
same-topology summary tree. Topology-varying posterior samples are
outside the current rateMap() workflow; supporting that
case would require a future extension that can summarize clades absent
from some sampled trees.
For ordinary bifrost outputs, shifts are placed at
nodes, so each branch has one fitted regime-rate value. With the default
summary = "branch", rateMap() matches each
target-tree branch to the corresponding input-tree branch by descendant
tip set. The matched input branch contributes its fitted regime rate for
that run, and those branch-level values are averaged across runs using
the requested weighting scheme. The target tree supplies the reference
topology, edge order, branch lengths, and plotting geometry for the
final map. Use summary = "interval" only for true
stochastic-map inputs where states can change within branches.
For posterior tree samples, weights = "equal" is usually
the appropriate choice: each retained tree is one posterior draw, so the
goal is to average over uncertainty in the underlying tree geometry
rather than to weight alternative threshold or parameter-sweep outcomes
by IC support. Keep uncertainty = TRUE to record how much
the matched branch rates vary across the tree sample.
The chunk below is a template for same-topology posterior tree samples. Because this worked example uses the same jaw-shape tree as the target, it does not show a separate plot.
# Replace this with an MCC tree, posterior mean chronogram, or other
# same-topology reference tree when analyzing posterior tree samples.
jaw_target_tree <- fish.tree
jaw_target_rate_map <- rateMap(
jaw_threshold_runs,
target_tree = jaw_target_tree,
weights = "equal",
summary = "branch",
uncertainty = TRUE,
progress = TRUE,
control = list(check = "topology")
)Practical Takeaways
- Holding
min_descendant_tips = 10fixed keeps the candidate-node filter constant whileshift_acceptance_thresholdchanges. - Equal weights summarize robustness across selected threshold settings; IC weights emphasize lower-IC threshold outcomes.
- For same-topology posterior tree samples with different branch
lengths, use an explicit target tree,
control = list(check = "topology"), branch summaries, and equal weights. - The default branch-level, log-rate-category map matches
bifrost’s node-shift model. - In
plot(x, ...),n_categoriessets the target number of automatic category bins;category_bin_method = "equal"andcategory_breaksgive more direct bin control; numeric manual breaks also set proportional category legend segment widths;ncolorscontrols continuous ramps. - Uncertainty maps identify branches where inferred log-rates are stable or threshold-sensitive.
Conclusion
The single-run jaw-shape analysis identifies one supported
multi-regime configuration, but it is not the only useful view of the
empirical signal. A threshold sweep keeps the biological sampling
constraint fixed and asks whether the inferred rate pattern survives
more permissive or more conservative shift acceptance.
rateMap() turns that sweep into maps and tables that
separate robust high-rate regions from threshold-sensitive ones. In this
jaw-shape sweep, the highest log-rate lungfish-rich region remains
stable across equal-weight and IC-weighted summaries, while several
moderate-rate branches are more sensitive to threshold and weighting
choices.
This empirical sensitivity analysis complements simulation-based model performance assessment. Simulations show how the method behaves when the truth is known; rate maps show how one dataset behaves across plausible analysis choices.
Appendix
A. Full IC Category Metadata
The main IC-weighted category table above is intentionally condensed
so it can be read next to the figure it explains. The full generated
table, including color_bin, lower,
upper, color, value_min, and
value_max, is written with the vignette artifacts as
rate_map_ic_categories.csv for readers who need the
complete display metadata.
B. Original-Scale Rates
The default log-rate scale is often the easier scale for comparing
multiplicative differences in fitted rates. Use log = FALSE
when you need the original fitted-rate units. Print or plot the
resulting object the same way as the log-rate maps above. The generated
summary table below keeps the rendered preview concrete: these
original-scale fitted-rate summaries remain strictly positive, and
values below one correspond to negative values on the default log-rate
maps.
# Recompute and plot equal-weight summaries on the original rate scale.
jaw_raw_rate_map <- rateMap(
jaw_threshold_runs,
weights = "equal",
uncertainty = TRUE,
log = FALSE,
progress = TRUE
)
plot(
jaw_raw_rate_map,
palette = "Viridis",
legend_title = "Mean fitted rate"
)| quantity | value |
|---|---|
| branches | 170 |
| minimum mean fitted rate | 8.07 x 10^-10 |
| median mean fitted rate | 1.90 x 10^-7 |
| maximum mean fitted rate | 1.11 x 10^-5 |
| branches with mean fitted rate below 1 | 170 |
log = FALSE branch
map. All rows summarize the plotted arithmetic mean fitted rates on the
original rate scale; the below-one count explains why the default
log-rate summaries can be negative without implying invalid fitted
rates.
C. Continuous Interval Maps
The branch-category default is the closest match for ordinary
bifrost output, because fitted bifrost shifts
occur at nodes. The package stores these results using stochastic-map
infrastructure, but the fitted map is not a simulated stochastic history
with additional state changes inside branches. For that reason, the main
summaries above use summary = "branch".
rateMap() can also use summary = "interval"
for inputs that really do change state within branches, such as
stochastic maps whose states can be mapped to numerical rates. In that
setting, interval mode slices each target-tree branch on a shared depth
grid and projects the stochastic-map segments onto those intervals. That
is useful for true within-branch histories, but it is less informative
for the jaw-shape bifrost sweep, where the interval map
would mostly restate the branch-level result with a different plotting
convention.
D. Branch Summary Table
For reporting, sort branches by their central log-rate estimate and carry along the uncertainty columns. The rendered table translates the top rows into descendant-tip counts and representative taxa. In this run, the highest log-rate whole-branch summaries point to the same lungfish-rich region highlighted above.
# Rank branch summaries by their equal-weight mean fitted log-rate.
branch_rates <- jaw_rate_map_equal$intervals
highest_log_rate_branches <- branch_rates[
order(branch_rates$value, decreasing = TRUE),
c(
"edge", "parent", "child", "clade_key",
"value", "mean", "median", "sd",
"q025", "q975", "highest_density_interval_low", "highest_density_interval_high", "n"
)
]
# Format clade identifiers for a compact reader-facing table.
highest_log_rate_branches$clade_key <- vapply(
highest_log_rate_branches$clade_key,
rate_map_format_clade_key,
character(1)
)
head(highest_log_rate_branches, 10)| edge | tips | example_taxa | value | sd | hdi_width |
|---|---|---|---|---|---|
| 13 | 9 | Griphognathus sculpta; Griphognathus whitei; … | -11.41 | 0.06431 | 0.1786 |
| 14 | 6 | Griphognathus sculpta; Griphognathus whitei; … | -11.41 | 0.06431 | 0.1786 |
| 15 | 4 | Griphognathus sculpta; Griphognathus whitei; … | -11.41 | 0.06431 | 0.1786 |
| 16 | 3 | Griphognathus sculpta; Griphognathus whitei; … | -11.41 | 0.06431 | 0.1786 |
| 17 | 2 | Griphognathus sculpta; Griphognathus whitei | -11.41 | 0.06431 | 0.1786 |
| 18 | 1 | Griphognathus sculpta | -11.41 | 0.06431 | 0.1786 |
| 19 | 1 | Griphognathus whitei | -11.41 | 0.06431 | 0.1786 |
| 20 | 1 | Rhynchodipterus elginensis | -11.41 | 0.06431 | 0.1786 |
| 21 | 1 | Robinsondipterus longi | -11.41 | 0.06431 | 0.1786 |
| 23 | 1 | Holodipterus gogoensis | -11.41 | 0.06431 | 0.1786 |
tips gives the descendant-tip count,
example_taxa lists representative descendants, and
hdi_width is the default 95% shortest-interval width.
Repeated values reflect a single high-rate region whose descendant
branches share very similar fitted rate histories across the threshold
sweep.
E. Weighting-Scheme Diagnostic
The main comparison between weighting schemes is visual: compare the equal-weight map from Part 1 with the IC-weighted map in Figure 1. This appendix section gives the code and table for a more mechanical diagnostic: compare equal-weight and IC-weighted branch summaries directly to find branches where the better-supported threshold outcomes emphasize different log-rates than the unweighted sensitivity average.
# Build parallel branch summaries under equal and IC weighting.
equal_branch_map <- rateMap(
jaw_threshold_runs,
summary = "branch",
weights = "equal",
progress = TRUE
)
ic_branch_map <- rateMap(
jaw_threshold_runs,
summary = "branch",
weights = "ic",
progress = TRUE
)
# Join corresponding branches and rank their absolute weighting differences.
comparison <- merge(
equal_branch_map$intervals[, c("edge", "value")],
ic_branch_map$intervals[, c("edge", "value")],
by = "edge",
suffixes = c("_equal", "_ic")
)
comparison$difference <- comparison$value_ic - comparison$value_equal
comparison$abs_difference <- abs(comparison$difference)
comparison <- comparison[order(comparison$abs_difference, decreasing = TRUE), ]
head(comparison, 10)| edge | tips | example_taxa | equal | ic | difference | abs_diff |
|---|---|---|---|---|---|---|
| 2 | 69 | Achoania jarvikii; Arquatichthys porosus; … | -22.3 | -25.79 | -3.488 | 3.488 |
| 139 | 16 | Austelliscus ferox; Cheirolepis jonesi; … | -22.3 | -25.79 | -3.488 | 3.488 |
| 1 | 85 | Achoania jarvikii; Arquatichthys porosus; … | -24.2 | -25.79 | -1.589 | 1.589 |
| 170 | 1 | Megamastax amblyodus | -24.2 | -25.79 | -1.589 | 1.589 |
| 3 | 66 | Arquatichthys porosus; Asperocephalus milleri; … | -16.65 | -15.06 | 1.586 | 1.586 |
| 134 | 3 | Achoania jarvikii; Guiyu oneiros; … | -16.65 | -15.06 | 1.586 | 1.586 |
| 135 | 2 | Achoania jarvikii; Psarolepis romeri | -16.65 | -15.06 | 1.586 | 1.586 |
| 136 | 1 | Achoania jarvikii | -16.65 | -15.06 | 1.586 | 1.586 |
| 137 | 1 | Psarolepis romeri | -16.65 | -15.06 | 1.586 | 1.586 |
| 138 | 1 | Guiyu oneiros | -16.65 | -15.06 | 1.586 | 1.586 |
tips and example_taxa summarize the descendant
clade; equal and ic are the two mean log-rate
summaries; difference is ic - equal; and
abs_diff is the absolute magnitude of that change.
Small differences suggest the broad branch log-rate picture is not very sensitive to weighting choice. Large differences deserve closer inspection, especially on branches central to the biological interpretation. Here the largest differences occur on a few moderate-rate clades and singleton branches; the highest log-rate lungfish-rich branches remain high under both weighting schemes.
Acknowledgements and Provenance
The rateMap() plotting layer follows the
phytools SIMMAP plotting ecosystem described in Part 1:
rate-valued summaries are drawn using the same lineage of tools as
densityMap(), plotSimmap(), and
plotTree().
References
- Clavel, J., Aristide, L., and Morlon, H. (2019). A Penalized Likelihood Framework for High-Dimensional Phylogenetic Comparative Methods and an Application to New-World Monkeys Brain Evolution. https://doi.org/10.1093/sysbio/syy045
- Otsu, N. (1979). A threshold selection method from gray-level histograms. IEEE Transactions on Systems, Man, and Cybernetics, 9(1), 62-66. https://doi.org/10.1109/TSMC.1979.4310076
- Paradis, E., and Schliep, K. (2019). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics, 35, 526-528. https://doi.org/10.1093/bioinformatics/bty633
- Revell, L. J. (2013). Two new graphical methods for mapping trait evolution on phylogenies. Methods in Ecology and Evolution, 4, 754-759.
- Revell, L. J. (2014). Graphical methods for visualizing comparative data on phylogenies. Chapter 4 in Modern Phylogenetic Comparative Methods and Their Application in Evolutionary Biology: Concepts and Practice, 77-103.
- Revell, L. J. (2024). phytools 2.0: an updated R ecosystem for phylogenetic comparative methods (and other things). PeerJ, 12, e16505. https://doi.org/10.7717/peerj.16505
- Troyer, E. M., et al. (2025). Macroevolutionary role reversals in the earliest radiation of bony fishes. https://doi.org/10.1016/j.cub.2025.08.008
Software Used in This Vignette
-
bifrostfor shift searches andrateMap()summaries. -
apeandphytoolsfor phylogenetic data structures, SIMMAP trees, and plotting.