Whole-Tree Models, PCA, and bifrost
Source:vignettes/pca-model-selection-and-bifrost-vignette.Rmd
pca-model-selection-and-bifrost-vignette.RmdIntroduction
This vignette builds on the Brownian-motion and covariance background
developed in Brownian
Motion and Multivariate Shifts. It considers how BM, OU, and EB
behave as whole-tree models, what happens when empirical datasets are
more heterogeneous than those single-process descriptions allow, and
where bifrost sits within that broader landscape.
Whole-tree models are useful because they provide simple ways to describe how variation accumulates across a phylogeny. But that simplicity can also become a limitation when different parts of the tree follow different local regimes, since a single fit may average over structure that is biologically important. PCA often enters this discussion as an appealing way to simplify multivariate data, yet its leading axes are chosen to maximize sample variance rather than preserve the evolutionary structure we necessarily care about. The goal here is to clarify what whole-tree models capture, what they can obscure, and why PCA is not a neutral substitute for modeling heterogeneity directly.
1. BM, OU, EB, and the limits of whole-tree models
| Model | Core idea | Main parameters | Typical biological reading |
|---|---|---|---|
| BM | Diffusion with no preferred direction and constant rate through time | Trait variance accumulates steadily as lineages diverge | |
| OU | Diffusion plus pull toward an optimum | , , | Traits wander, but are restrained around an adaptive peak |
| EB | Diffusion with a systematic tempo change through time | , | Evolution is fastest early and then slows, or more generally speeds up or slows down through time |
Under BM, variance accumulates linearly with time and no optimum is
preferred. That makes BM a useful baseline for asking whether a
multivariate rate structure is constant across a tree. When
bifrost searches for branch-specific shifts under BM/BMM,
it is still working inside that diffusion-based family.
OU changes the story by adding deterministic pull toward an optimum. The parameter controls how strongly lineages are drawn back toward , so OU is often read as a simple model of adaptive constraint or stabilization (Hansen 1997; Butler and King 2004). EB is different again: it is not about attraction to an optimum, but about a directional change in tempo through time, often written as a rate like , with classic early-burst behavior corresponding to (Harmon et al. 2010).
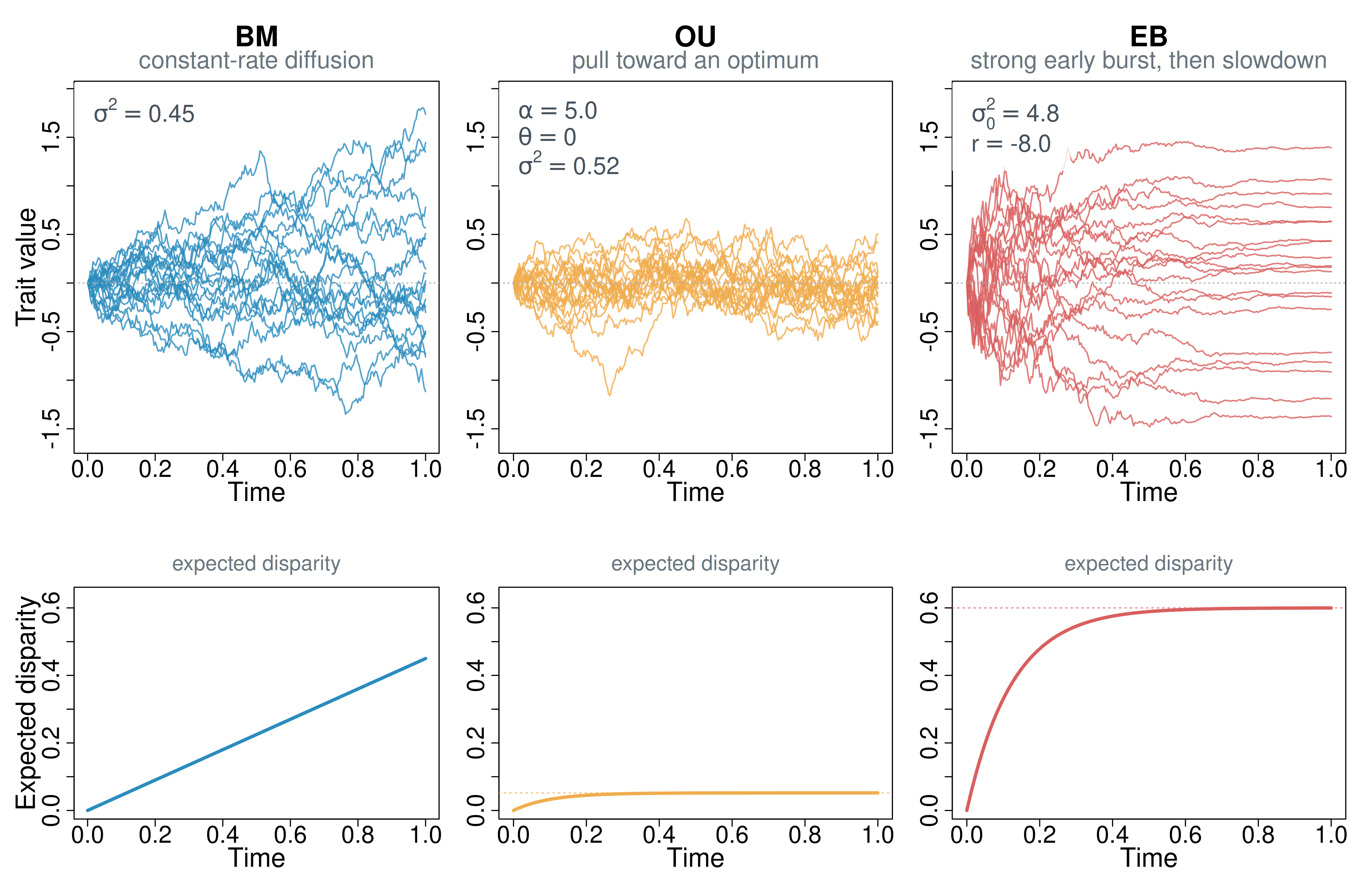
Figure 1. Three simple continuous-trait models contrasted with simulations and their theoretical disparity implications. Top row: simulated trajectories under BM, OU, and EB. BM diffuses at a constant rate, so trajectories spread steadily through time. OU adds deterministic pull toward an optimum, which keeps trajectories more tightly bounded around the center. EB begins with a stronger early burst and then slows sharply, so most divergence is front-loaded. Bottom row: the corresponding theoretical expected disparity-through-time curves under the same parameter values. BM increases linearly, OU rises and saturates, and EB accumulates disparity early before flattening.
Figure 1 also clarifies the scope of these models. BM, OU, and EB each provide a coarse whole-tree summary of how variation accumulates: BM assumes one diffusion process across the tree, OU adds one global pull structure, and EB allows one simple tree-wide trend in tempo. If a dataset instead contains patchy clade-specific shifts, mixtures of slower and faster regimes, or several distinct episodes of change, then even these more flexible whole-tree descriptions can miss the signal. EB is the clearest example: it allows non-uniformity, but only as one smooth tree-wide tempo trend rather than as localized shifts.
2. When the process is heterogeneous across the tree
That kind of mismatch is where bifrost becomes useful.
The package does not replace BM with a different global family. Instead,
it keeps the local process Brownian and allows different parts of the
tree to occupy different multivariate BM/BMM regimes. In that sense,
variation across an empirical tree can be approximated as a multi-regime
Gaussian/Brownian process on a painted phylogeny: within each regime,
traits still evolve as multivariate Gaussian diffusion, but across the
tree the observed pattern can reflect several branch- or clade-specific
regimes rather than one uniform model for the entire dataset.
This matters because heterogeneity is not only a problem for whole-tree fits. If the underlying process is patchy, nested, or recurrent across the phylogeny, then even models assigned to large clades can still average over mixed local regimes and provide a poor description of the data. Figure 2 extends the simpler descendant-clade shift logic introduced in Brownian Motion and Multivariate Shifts to that more heterogeneous setting.
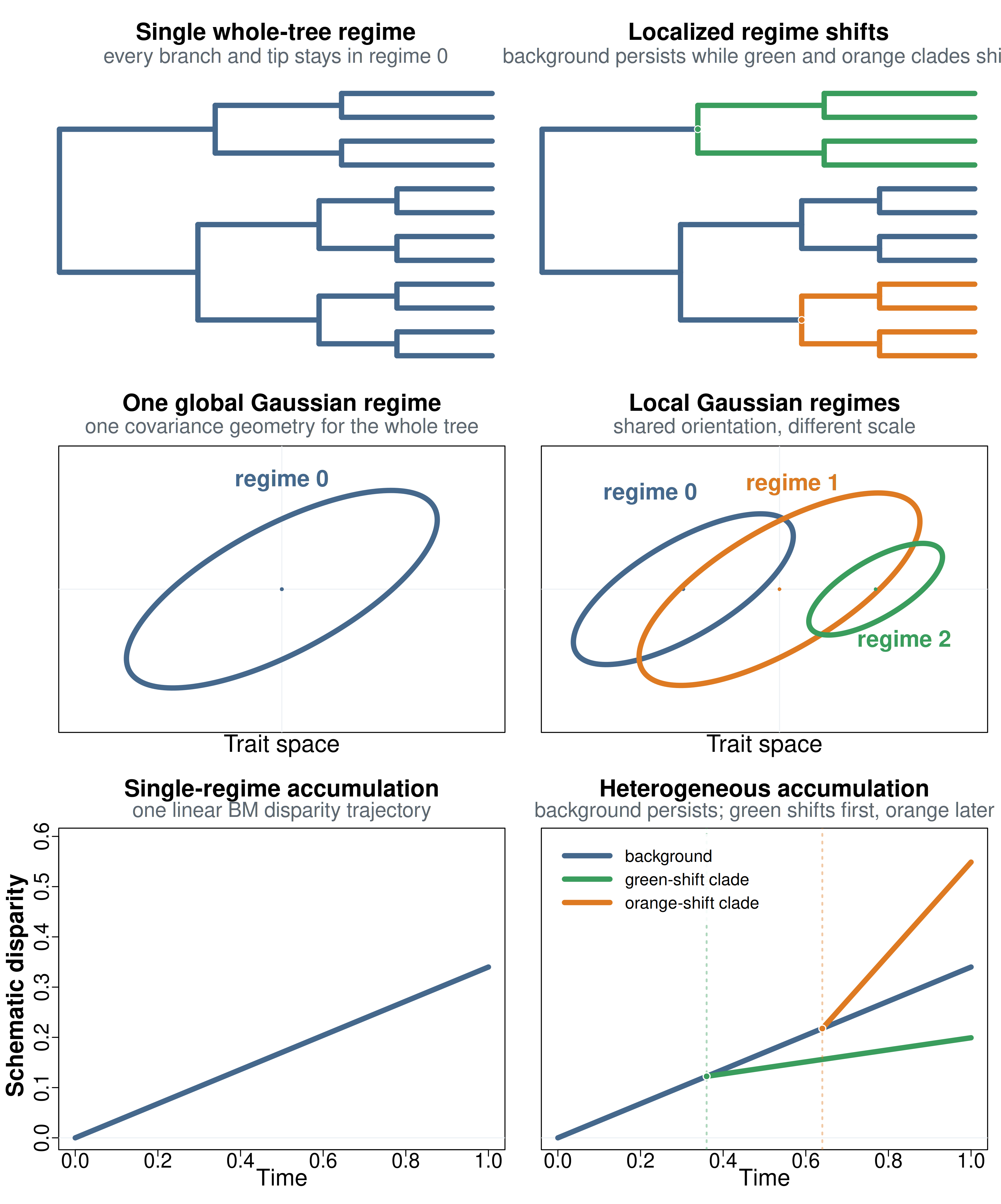
Figure 2. Whole-tree summaries versus heterogeneous
regime mixtures. Left column: one global BM/BMM regime applies
everywhere, yielding one tree-wide Gaussian trait-space geometry and one
linear disparity-accumulation pattern. Right column: localized
clade-specific shifts create multiple regimes across the tree; under
bifrost’s current assumptions those local regimes remain
multivariate Gaussian with shared covariance geometry but different
scale, and their disparity accumulation patterns can diverge after
shifts. Horizontal offsets in the trait-space panel are only for visual
separation, not different regime means. Those local trajectories need
not be well summarized by any single whole-tree fit.
This perspective is close in spirit to mixed Gaussian phylogenetic
model frameworks such as PCMFit, which also allow different
Gaussian evolutionary descriptions to be assigned to different parts of
a tree and search among alternative shift configurations (Mitov, Bartoszek, and
Stadler 2019; Mitov et al. 2020).
The main distinction is that bifrost is currently narrower
and more directly oriented toward high-dimensional original trait data.
It keeps the local process within the multivariate BM/BMM family, fits
regime structure directly in trait space through penalized multivariate
GLS, and assumes proportional regime scaling rather than the broader
class of Gaussian shift models considered in PCMFit.
Because bifrost fits heterogeneous regime structure
directly, it can capture much more complex rate-shift patterns than a
single whole-tree BM, OU, or EB fit while still remaining inside a
Brownian, covariance-based framework.
bayou is another nearby comparison (Uyeda and Harmon 2014),
but it addresses a different kind of heterogeneity: OU adaptive-regime
shifts rather than the multivariate BM/BMM rate shifts targeted by
bifrost. Multivariate OU models remain an important area of
method development (Mariadassou, Lambert, and
Morlon 2018), whereas bifrost’s current niche is
heterogeneous multivariate BM/BMM with branch-level rate shifts under
proportional regime scaling.
3. PCA Is Not a Neutral Fix
When multivariate data are high-dimensional and difficult to summarize directly, one common response is to simplify them first, usually with PCA, rather than model the heterogeneity itself. That can be useful for visualization and exploratory summaries, but it is not a neutral preprocessing step for comparative inference (Revell 2009; Uyeda, Caetano, and Pennell 2015). PCA can hide the axes carrying the biologically relevant difference, and it can also reorder axes in ways that change which global model looks best supported (Uyeda, Caetano, and Pennell 2015).
Why PCA Truncation Can Mislead
The first of those problems is geometric. PCA orders axes by realized sample variance, not by whether they contain the biologically relevant contrast. If the process of interest lies partly along a lower-variance direction, then truncating to the leading PC can discard the very signal we want to interpret, a point long recognized more generally in the PCA literature (Jolliffe 1982). This is easiest to see in a simple two-regime example where most variation lies along one axis, but the important difference lies along another.
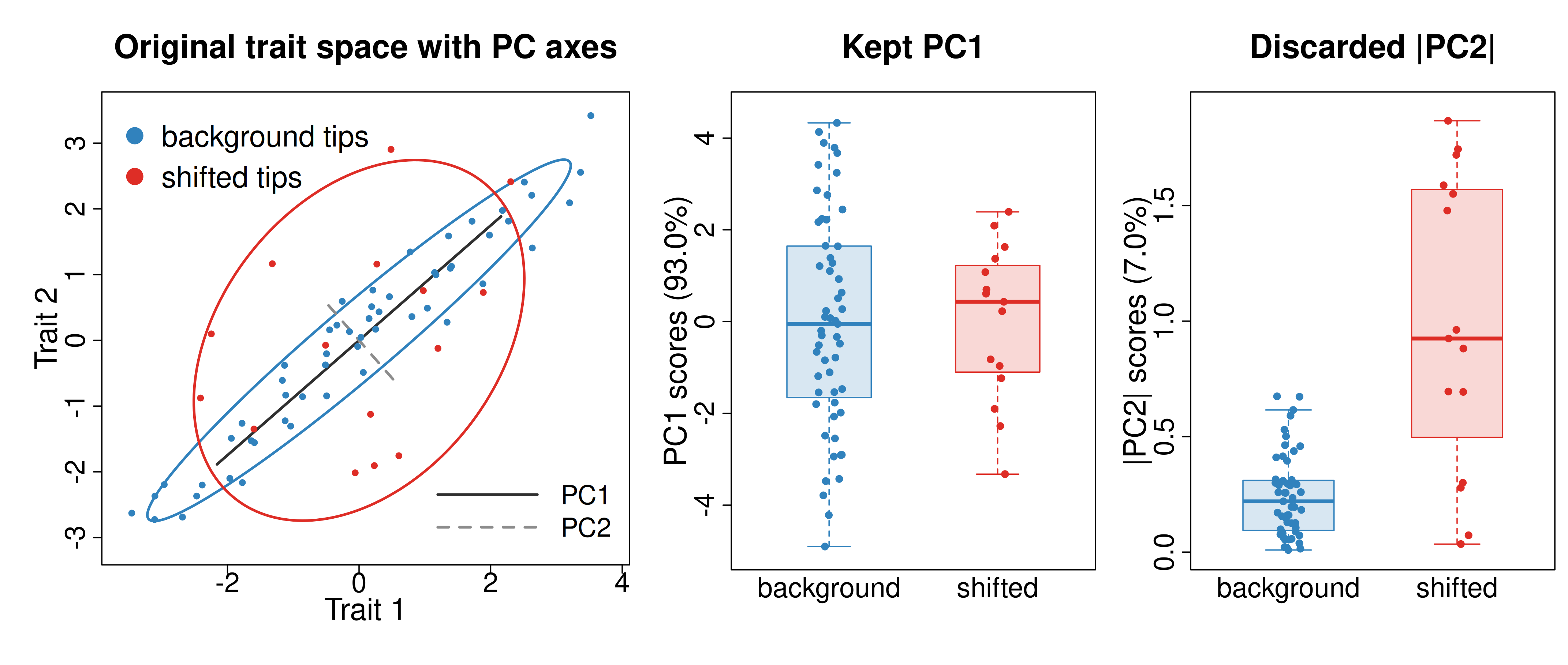
Figure 3. Why truncating PCA can mislead. Left: both regimes share the same broad diagonal axis of variation, but the shifted regime has much greater spread perpendicular to that axis; the overlaid lines show PC1 and PC2. Middle: scores on retained PC1 overlap strongly even though PC1 captures most total variance. Right: the regime difference is clear on the discarded low-variance axis, shown here as absolute distance on PC2 because the difference is in spread rather than mean.
Here both regimes share the same broad major axis, but the shifted
regime has much greater variance along the lower-variance axis
perpendicular to it. PCA still assigns 93.0% of the total sample spread
to PC1 because that long diagonal axis dominates the variance budget.
Yet the retained PC1 scores overlap strongly, while the discarded
|PC2| scores are exactly where the regime difference
becomes clear. Because the shift is in spread rather than mean, the
contrast appears in |PC2| rather than in signed PC2
scores.
Phylogenetic PCA can be more coherent than ordinary PCA under a Brownian-motion null, but it is still a model-based rotation rather than a neutral preprocessing step (Revell 2009; Uyeda, Caetano, and Pennell 2015).
PCA Can Distort Model Selection
The truncation problem above is only one PCA caution. A second issue is more inferential: PCA can change which comparative model appears best supported. In the simulations of Uyeda, Caetano, and Pennell (2015), even when all traits truly evolved under constant-rate multivariate BM, the first few standard PC axes often looked as though they were better fit by an early-burst model. The point is not that PCA invents an early burst out of thin air; it is that ranking axes by realized sample variance across the whole tree can privilege combinations of traits with unusually large deep divergences.
We show a compact illustration of this result using a reduced version of the equal-rate BM setup discussed in Uyeda, Caetano, and Pennell (2015): 20 replicate pure-birth trees with 50 tips, 20 independent BM traits, and BM, OU, and EB model fits to both the original traits and the first six standard PC scores.
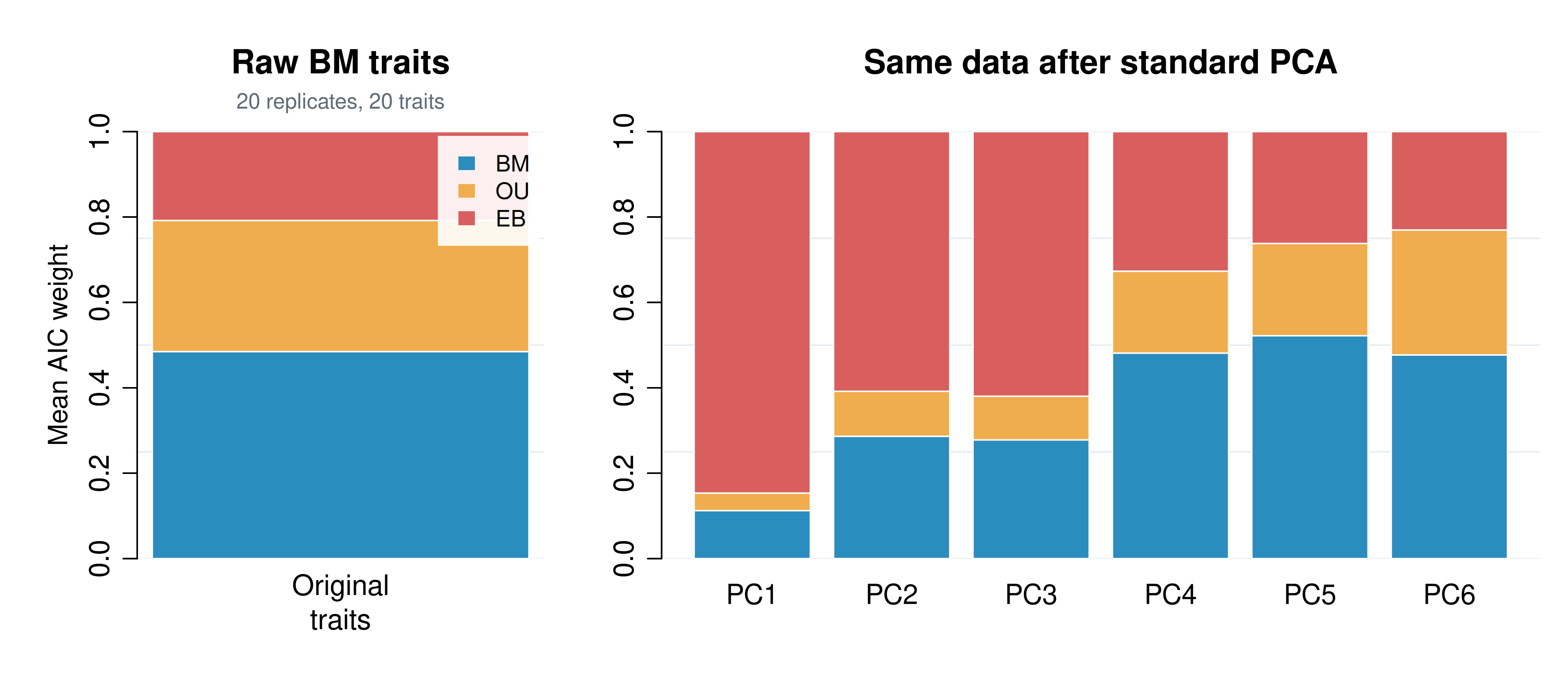
Figure 4. A compact illustration of the standard-PCA artifact emphasized by Uyeda, Caetano, and Pennell (2015). Left: averaged across the original traits, BM has the highest support. Right: after standard PCA, support is pulled toward EB on the earliest axes even though the generating process was BM throughout.
The PCA issue here is different from the truncation problem above. The signal is not being hidden on a low-variance axis. Instead, standard PCA changes which univariate combinations get placed first. Averaged across the original BM traits, BM remains the best-supported model, although its mean AIC weight does not approach 1 because OU and EB collapse onto BM when their extra parameter is unnecessary; under this three-model comparison, the BM ceiling is only about 0.58, as Uyeda et al. discuss. After standard PCA, the leading axes are no longer a neutral sample from the multivariate process: they are the axes most likely to make a constant-rate BM data set look EB-like.
PCA can be useful descriptively, but once the transformed axes are
treated as the units of model comparison, the ranking itself can change
which macroevolutionary explanations look plausible. PCA is a
variance-maximizing dimension reduction, not a model of tree-structured
heterogeneity. It therefore does not rescue a heterogeneous dataset from
the limitations of a single whole-tree fit; it can add another layer of
distortion on top of that original mismatch. That is why
bifrost fits models in the original trait space and treats
PCA as optional description rather than as primary preprocessing for
inference.
4. Mapping Theory to the bifrost API
With that in mind, the main package interface becomes easier to read.
bifrost is most useful when a single whole-tree summary is
too crude, but a Brownian, covariance-based description is still a
reasonable local approximation within regimes. Relative to broader
mixed-Gaussian frameworks such as PCMFit, it trades model
generality for tractability in high-dimensional original trait
space.
bifrost handles high-dimensional trait data through
penalized-likelihood multivariate GLS fits implemented in
mvMORPH::mvgls (Clavel, Aristide, and
Morlon 2019). In ordinary multivariate GLS, covariance estimation
becomes unstable or singular when the number of traits is large relative
to the number of species. Penalization regularizes those covariance
estimates, which makes it possible to work directly in the original
trait space rather than collapsing the data to a few PCs first. In
bifrost, that high-dimensional machinery is combined with
proportional regime scaling, which further keeps the BM/BMM search
tractable.
| Theoretical idea |
bifrost component |
Interpretation |
|---|---|---|
| Single-regime baseline process |
baseline_tree, formula,
baseline_ic
|
One multivariate BM model shared across the tree |
| Candidate evolutionary shift | internal nodes passing
min_descendant_tips
|
A descendant clade is allowed to have a different regime |
| Model comparison |
IC,
shift_acceptance_threshold, optimal_ic
|
Controls how much better a more complex model must fit |
| Regime-specific rate structure | VCVs |
Estimated regime-specific variance-covariance matrices, interpreted in the current model through proportional scaling across regimes |
| Support for accepted shifts | ic_weights |
Drop-one support values for shifts in the final model |
| Search dynamics | model_fit_history |
Sequence of accepted and rejected candidates |
library(bifrost)
fit <- searchOptimalConfiguration(
baseline_tree = tree,
trait_data = trait_data,
formula = "trait_data ~ 1",
min_descendant_tips = 10,
num_cores = 4,
shift_acceptance_threshold = 20,
uncertaintyweights_par = TRUE,
IC = "GIC",
method = "H&L",
error = TRUE,
plot = FALSE
)
fit$shift_nodes_no_uncertainty
fit$VCVs
fit$ic_weightsFor intercept-only morphometric datasets,
formula = "trait_data ~ 1" corresponds to the multivariate
BM/BMM case discussed in Part 1, Brownian Motion and
Multivariate Shifts. At present, that means BM/BMM fits with
proportional regime scaling rather than arbitrary covariance
reorientation across clades. For multivariate pGLS-style analyses with
predictors, the same logic applies except that the covariance model now
describes residual structure after accounting for the predictors.
Practical Takeaways
- BM, OU, and EB are useful whole-tree summaries, but they can fit heterogeneous datasets poorly when local regimes are nested, patchy, or clade-specific.
-
bifrostaddresses that problem by fitting heterogeneous multivariate BM/BMM regimes directly in the original trait space, using penalized multivariate GLS to remain tractable for high-dimensional data. - PCA is useful descriptively, but its axes maximize sample variance rather than preserve the evolutionary structure we necessarily care about.
- PCA can mislead inference in more than one way: truncation can discard biologically relevant low-variance structure, and axis ranking can make different comparative models look spuriously attractive.
-
bifrosttherefore treats PCA as optional description rather than required preprocessing, and its current niche is multivariate BM/BMM shift detection under proportional regime scaling.
Next Steps
If you want to connect this back to the broader theoretical setup or a worked analysis:
- Part 1, Brownian Motion and Multivariate Shifts, lays out the BM and covariance ideas assumed here;
- the jaw-shape vignette shows a full morphometric workflow on a real dataset.
References
- Butler, M. A., and King, A. A. (2004). Phylogenetic Comparative Analysis: A Modeling Approach for Adaptive Evolution. https://doi.org/10.1086/426002
- Clavel, J., Aristide, L., and Morlon, H. (2019). A Penalized Likelihood Framework for High-Dimensional Phylogenetic Comparative Methods and an Application to New-World Monkeys Brain Evolution. https://doi.org/10.1093/sysbio/syy045
- Hansen, T. F. (1997). Stabilizing Selection and the Comparative Analysis of Adaptation. https://doi.org/10.1111/j.1558-5646.1997.tb01457.x
- Harmon, L. J., Losos, J. B., Davies, T. J., Gillespie, R. G., Gittleman, J. L., Jennings, W. B., Kozak, K. H., McPeek, M. A., Moreno-Roark, F., Near, T. J., Purvis, A., Ricklefs, R. E., Schluter, D., Schulte II, J. A., Seehausen, O., Sidlauskas, B. L., Torres-Carvajal, O., Weir, J. T., and Mooers, A. O. (2010). Early Bursts of Body Size and Shape Evolution Are Rare in Comparative Data. https://doi.org/10.1111/j.1558-5646.2010.01025.x
- Jolliffe, I. T. (1982). A Note on the Use of Principal Components in Regression. https://doi.org/10.2307/2348005
- Mariadassou, M., Lambert, A., and Morlon, H. (2018). Inference of Adaptive Shifts for Multivariate Correlated Traits. https://doi.org/10.1093/sysbio/syy005
- Mitov, V., Bartoszek, K., and Stadler, T. (2019). Automatic Generation of Evolutionary Hypotheses Using Mixed Gaussian Phylogenetic Models. https://doi.org/10.1073/pnas.1813823116
- Mitov, V., Bartoszek, K., Asimomitis, G., and Stadler, T. (2020). Fast Likelihood Calculation for Multivariate Gaussian Phylogenetic Models with Shifts. https://doi.org/10.1016/j.tpb.2019.11.005
- Revell, L. J. (2009). Size-Correction and Principal Components for Interspecific Comparative Studies. https://doi.org/10.1111/j.1558-5646.2009.00804.x
- Uyeda, J. C., and Harmon, L. J. (2014). A Novel Bayesian Method for Inferring and Interpreting the Dynamics of Adaptive Landscapes from Phylogenetic Comparative Data. https://doi.org/10.1093/sysbio/syu057
- Uyeda, J. C., Caetano, D. S., and Pennell, M. W. (2015). Comparative Analysis of Principal Components Can Be Misleading. https://doi.org/10.1093/sysbio/syv019
Software Used in This Vignette
- Ho, L. S. T., and Ané, C. (2014). A Linear-Time Algorithm for
Gaussian and Non-Gaussian Trait Evolution Models. Systematic
Biology, 63(3), 397-408. [
phylolm] - Clavel, J., Escarguel, G., and Merceron, G. (2015). mvMORPH: an R package for fitting multivariate evolutionary models to morphometric data. Methods in Ecology and Evolution, 6(11), 1311-1319. https://doi.org/10.1111/2041-210X.12420
- Paradis, E., and Schliep, K. (2019). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics, 35, 526-528. https://doi.org/10.1093/bioinformatics/bty633
- Revell, L. J. (2024). phytools 2.0: an updated R ecosystem for phylogenetic comparative methods (and other things). PeerJ, 12, e16505. https://doi.org/10.7717/peerj.16505