
Phylogeny is the conceptual and practical framework for studying biodiversity from an evolutionary perspective. My research program unifies hypothesis testing with data-driven approaches in phylogenetics to uncover the factors that generate evolutionary patterns, a concept described as “Phylogenetic Natural History” (Uyeda et al. 2018).
Phylogeny Inference
Resolving the evolutionary history of groups that rapidly diversified remains a fundamental challenge in systematic biology. My interests in this area are motivated by the extinction of non-avian dinosaurs and the subsequent diversification of vertebrate clades around the K-Pg boundary (~66 Ma).

In one flagship example, we investigated relationships among modern bird families and estimated a new timescale of avian diversification (Prum, Berv, et al. 2015, Nature). We leveraged target-capture DNA sequencing to infer nine super-ordinal groups of modern birds and advanced approaches for phylogenetic experimental design, including phylogenetic informativeness metrics for improving study power.
Download Paper Download Poster
View poster inline
Divergence Time Estimation
A longstanding issue in systematic biology is conflict between inferred ages from fossils versus molecular sequences. For birds, fossils often imply key divergences close to the K-Pg boundary (~66 Ma), while molecular clocks can suggest substantially older ages.
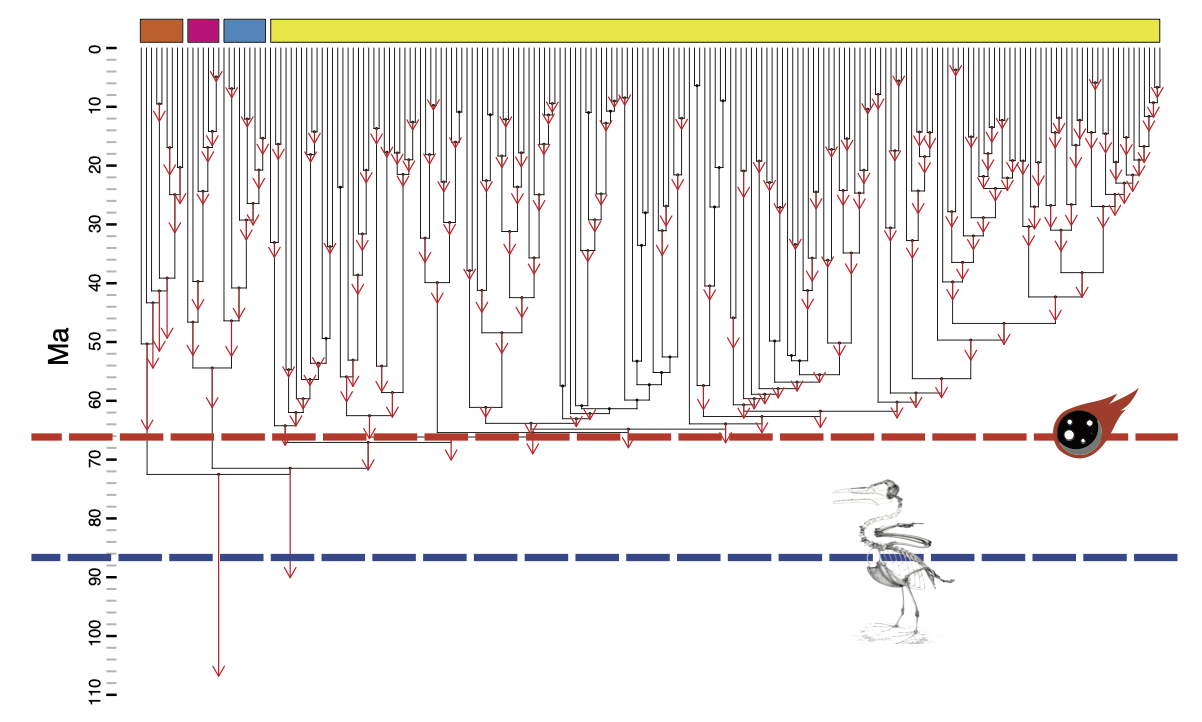
In Berv and Field 2018, Systematic Biology, we tested whether a K-Pg-associated acceleration of avian genomic evolution contributes to this discrepancy. The hypothesis builds on the “Lilliput effect”: if mass extinctions favor smaller-bodied taxa and smaller birds evolve faster genomes, molecular clocks may systematically overestimate ages for major avian divergences.
Download Paper 2019 Defense Seminar Publisher's Award
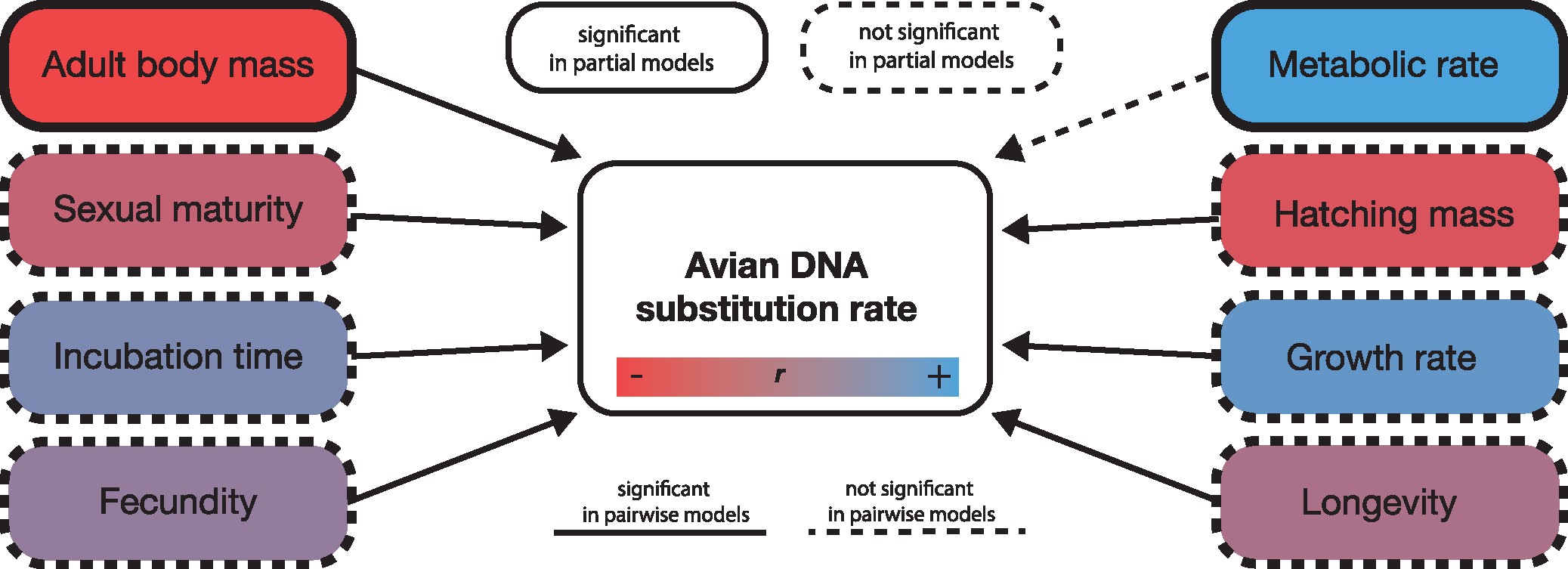
Tempo and Mode of Molecular Evolution
As a Life Sciences Fellow at the University of Michigan (2019-2023), I explored how variation in both evolutionary tempo (rate) and mode (substitution process) can confound phylogenetic inference.
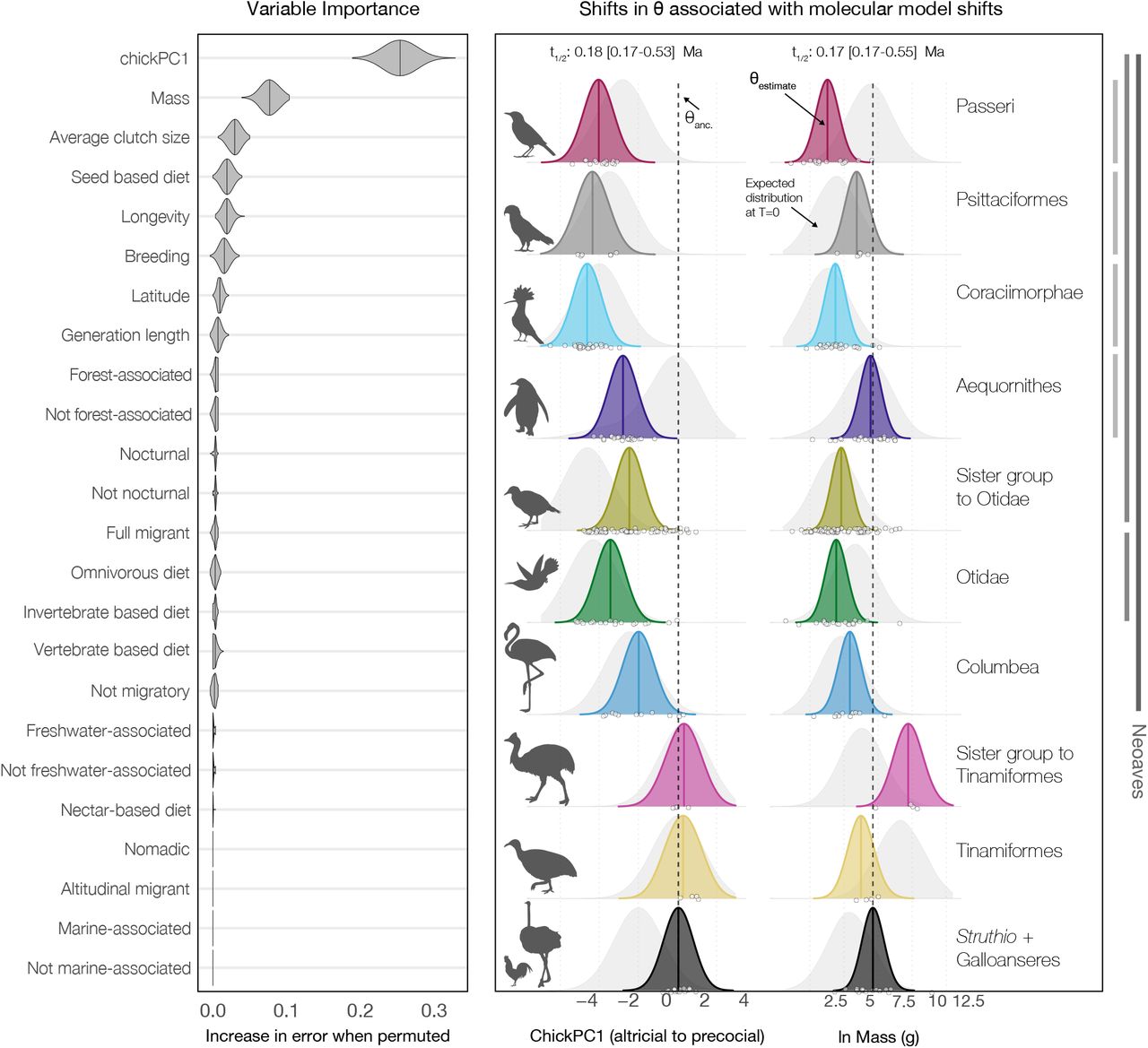
In Berv et al. 2024 (Science Advances), we tested how these processes shaped early crown-bird history using a phylogenomic dataset of 198 avian lineages and 910 loci across exons, introns, untranslated regions, and mitochondrial genomes.
These analyses were implemented with janus, our framework for detecting shifts in molecular substitution models and base composition across phylogenies (see janus on the Software page).
We inferred 17 molecular model shifts across 12 phylogenetic edges, with 15 concentrated within ~5 million years of the K-Pg boundary. Shift locations were strongly associated with developmental mode, adult body mass, and metabolic allometry, supporting an integrated post-extinction reorganization of avian genomes and life-history evolution.
Read in Science Advances Download PDF Janus software card Janus repository
Habitat-Associated Molecular Evolution in the Deep Sea
In Evolution (2025), Amelia Weiss and I tested whether habitat longevity predicts rates of molecular evolution in deep-sea specialists. Using mitochondrial CO1 data from bathymodioline mussels and siboglinid tubeworms, we compared lineages occupying organic falls, hydrothermal vents, and cold seeps.
Read in Evolution Download PDF Data + code
Watch short explainer video
Organic falls, hydrothermal vents, and cold seeps persist over dramatically different timescales (from decades to millennia), imposing hard limits on ecological persistence and selecting for distinct life-history strategies.
Key findings:
- Substitution rates show an inverse relationship with habitat longevity.
- Organic-fall specialists evolve fastest; vent and seep lineages are slower.
- After phylogenetic correction, body size does not explain rate differences.

Because the same pattern appears in two deeply diverged phyla, the results support a broader resource-longevity framework: ecological persistence can shape life-history strategy and the tempo of molecular evolution.
